
探究IgM型多发性骨髓瘤(MM)患者的临床特征、疗效及预后。
收集中国医学科学院血液病医院2009年12月18日至2020年10月29日收治的6例IgM型MM患者临床资料,对其临床特点、实验室检查结果、骨髓检查结果、疗效及预后进行总结、分析。
6例患者均符合IgM型MM的诊断标准。男4例,女2例,初次诊断中位年龄为70(59~81)岁。Durie-Salmon(DS)分期:ⅠA期2例,ⅢA期4例;国际分期系统(ISS)分期:Ⅱ期4例,Ⅲ期2例。初发症状:骨痛4例,高黏滞血症3例,淋巴结肿大或肝脾大2例。实验室检查:中位血清M蛋白39.11(3.61~75.56)g/L,中位血清IgM 69.35(4.35~137.00)g/L,中位HGB 87(70~131)g/L,中位血清肌酐83.6(53.0~129.6)μmol/L,中位血清钙2.12(2.11~2.50)mmol/L。中位骨髓浆细胞比例为0.390(0.255~0.590),4例患者外周血细胞形态学可见浆细胞。染色体核型分析:低二倍体1例。FISH检查:2例P53基因缺失,1例1q21扩增阳性,4例RB-1基因缺失阳性;6例患者IgH重排均阳性,其中3例CCND1/IgH融合基因阳性,明确为t(11;14)重排改变。免疫表型:CD38、CD138、单克隆轻链表达均阳性,4例CD20个别阳性。6例患者均接受蛋白酶体抑制剂为主的方案治疗,达到部分缓解至严格意义的完全缓解,治疗敏感性尚可。
IgM型MM除骨髓瘤典型临床表现外,可伴高黏滞血症、淋巴结肿大或肝脾大等特征性表现,细胞遗传学以t(11;14)多见治疗反应及预后与常见骨髓瘤亚型无明显区别。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
多发性骨髓瘤(multiple myeloma,MM)是一种以浆细胞异常增殖为特征的血液系统恶性肿瘤,分泌大量单克隆免疫球蛋白为其主要特征[1],其中分泌IgM的MM非常罕见,仅占0.5%~0.8%[2,3],其临床特征及预后不详,国内尚未见大系列报道。本文对我中心6例IgM型MM患者进行回顾性分析及文献复习,以了解IgM型MM患者的临床特征、疗效及预后。
入选病例为2009年12月18日至2020年10月29日就诊于中国医学科学院血液病医院的6例IgM型MM患者。所有患者均符合文献[4]的诊断标准,临床分期均按照Durie-Salmon(DS)分期和国际分期系统(ISS)标准。
6例患者均进行G显带核型分析和FISH检查。骨髓标本均采用德国美天旎生物技术有限公司抗CD138磁珠进行浆细胞分选、富集(纯度>90%),应用DNA探针(美国Abbott公司产品)间期FISH(iFISH)技术检测以下改变:1q21扩增、RB1缺失、13q14.3缺失、IgH重排、P53缺失,IgH重排阳性者进行t(4;14)、t(11;14)、t(14;16)、t(14;20)检查。
疗效评价标准参照文献[5]。随访截止时间为2021年1月30日,中位随访时间18.1(3.1~39.6)个月。随访采用查阅患者住院病历和电话随访的形式。5例患者完成随访,1例患者因无法联系失访。
无进展生存(PFS)期定义为MM诊断至疾病进展、复发或死亡的时间;总生存(OS)期定义为MM诊断至死亡或随访截止时间。
患者实验室检查数据资料以中位数(范围)进行描述。
本组6例患者中,男4例,女2例,诊断中位年龄为70(59~81)岁。3例轻链为κ型,3例轻链为λ型,DS分期:ⅠA期2例,ⅢA期4例;ISS分期:Ⅱ期4例,Ⅲ期2例。
骨痛4例,高黏滞血症3例,淋巴结肿大或肝脾大2例。
贫血4例,中位HGB为87(70~131)g/L;白细胞减少1例,中位WBC为5.35(2.59~10.79)×109/L;血小板减少1例,中位PLT为222(30~431)×109/L;肾功能损害2例,中位血清肌酐83.6(53.0~129.6)μmol/L;血清钙水平均正常,中位血清钙2.12(2.11~2.50)mmol/L;中位血清M蛋白39.11(3.61~75.56)g/L;中位血清IgM为69.35(4.35~137.00)g/L,IgG为5.67(2.91~8.89)g/L,IgA为0.46(0.20~1.47)g/L,κ链6.38(2.65~16.22)g/L,λ链2.08(0.04~68.00)g/L。中位β2-微球蛋白水平为4.33(2.86~7.75)mg/L。患者具体临床资料见表1。
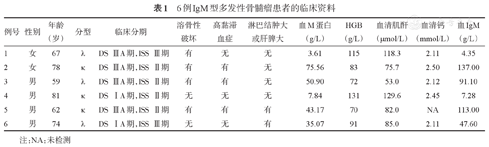
6例IgM型多发性骨髓瘤患者的临床资料
6例IgM型多发性骨髓瘤患者的临床资料
| 例号 | 性别 | 年龄(岁) | 分型 | 临床分期 | 溶骨性破坏 | 高黏滞血症 | 淋巴结肿大或肝脾大 | 血M蛋白(g/L) | HGB(g/L) | 血清肌酐(μmol/L) | 血清钙(mmol/L) | 血IgM(g/L) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 女 | 67 | λ | DS ⅢA期,ISS Ⅲ期 | 有 | 无 | 无 | 3.61 | 115 | 118.3 | 2.11 | 4.35 |
| 2 | 女 | 78 | κ | DS ⅢA期,ISS Ⅱ期 | 有 | 有 | 无 | 75.56 | 83 | 75.7 | 2.50 | 137.00 |
| 3 | 男 | 59 | λ | DS ⅢA期,ISS Ⅱ期 | 有 | 有 | 无 | 50.90 | 72 | 53.0 | 2.12 | 91.10 |
| 4 | 男 | 81 | κ | DS ⅠA期,ISS Ⅱ期 | 无 | 无 | 无 | 7.84 | 131 | 129.6 | 2.45 | 7.28 |
| 5 | 男 | 62 | κ | DS ⅢA期,ISS Ⅱ期 | 有 | 有 | 有 | 43.17 | 70 | 82.0 | NA | 113.00 |
| 6 | 男 | 74 | λ | DS ⅠA期,ISS Ⅲ期 | 无 | 无 | 有 | 35.07 | 91 | 85.0 | 2.11 | 47.60 |
注:NA:未检测
6例患者骨髓细胞形态学均可见浆细胞明显增多,中位浆细胞比例为0.390(0.255~0.590),2例患者可见浆样淋巴细胞增多。4例患者外周血细胞形态学可见浆细胞,中位外周血浆细胞比例0.01(0~0.03)。
染色体核型分析示正常核型5例,低二倍体1例。6例患者均进行FISH检查,2例P53基因缺失阳性,1例1q21扩增阳性(拷贝数为3,阳性率86%),4例RB-1基因缺失阳性。6例患者IgH重排均阳性,其中3例CCND1/IgH融合基因阳性,明确为t(11;14)重排改变。6例患者MYD88 L256P基因突变检测均阴性。
6例患者CD38、CD138、单克隆轻链表达均阳性,为异常浆细胞典型表型。4例患者CD20个别阳性。因例数较少,无法判断有无特殊抗原表达。骨髓检查具体结果见表2。
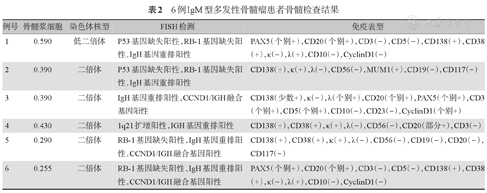
6例IgM型多发性骨髓瘤患者骨髓检查结果
6例IgM型多发性骨髓瘤患者骨髓检查结果
| 例号 | 骨髓浆细胞 | 染色体核型 | FISH检测 | 免疫表型 |
|---|---|---|---|---|
| 1 | 0.590 | 低二倍体 | P53基因缺失阳性,RB-1基因缺失阳性,IgH基因重排阳性 | PAX5(个别+),CD20(个别+),CD3(-),CD5(-),CD138(+),CD38(+),κ(-),λ(+),CD10(-),CyclinD1(-) |
| 2 | 0.390 | 二倍体 | P53基因缺失阳性,RB-1基因缺失阳性,IgH基因重排阳性 | CD138(+),κ(+),λ(-),CD56(-),MUM1(+),CD19(-),CD117(-) |
| 3 | 0.390 | 二倍体 | IgH基因重排阳性,CCND1/IGH融合基因阳性 | CD138(少数+),κ(-),λ(个别+),CD20(个别+),PAX5(个别+),CD3(个别+),CD5(个别+),CD10(-),CD23(-),CyclinD1(个别+) |
| 4 | 0.430 | 二倍体 | 1q21扩增阳性,IGH基因重排阳性 | CD138(+),CD38(+),κ(+),λ(-),CD56(-),CD20(部分+),CD3(-) |
| 5 | 0.290 | 二倍体 | RB-1基因缺失阳性,IgH基因重排阳性,CCND1/IGH融合基因阳性 | CD138(+),CD38(+),κ(+),λ(-),CD56(-),CD19(-),CD20(-),CD117(-) |
| 6 | 0.255 | 二倍体 | RB-1基因缺失阳性,IgH基因重排阳性,CCND1/IGH融合基因阳性 | PAX5(个别+),CD20(个别+),CD3(-),CD5(-),CD138(+),CD38(+),κ(-),λ(+),CD10(-),CyclinD1(-) |
所有患者均接受以蛋白酶体抑制剂为主的化疗方案。其中2例采用BCD方案(硼替佐米+环磷酰胺+地塞米松),2例采用IRD方案(伊沙佐米+来那度胺+地塞米松),1例入组临床试验,采用CMP方案(卡非佐米+美法仑+泼尼松),1例采用BD方案(硼替佐米+地塞米松)。6例患者经过诱导治疗后均至少达到部分缓解(PR),2例达到非常好的部分缓解(VGPR),1例达严格意义的完全缓解(sCR),治疗敏感性尚可。随访时间截至2021年1月30日,5例患者可获得完整随访数据,3例后期出现疾病进展而死亡,2例仍维持深度缓解。
MM是一种以分泌单克隆免疫球蛋白为特征的恶性浆细胞疾病,常见症状包括相关器官功能损伤,即"CRAB"症状(高钙血症、肾功能不全、贫血、骨病)。而高黏滞血症是MM相对罕见的并发症,但在IgM型MM中较常见,需引起注意。本组有3例(50%)患者初诊时即出现头晕、视物模糊、耳鸣等高黏滞血症表现,可能与患者血清IgM水平较高有关(3例均>90 g/L)。同时,IgM的相对分子质量为950×103,较其他类型免疫球蛋白(如IgG、IgA等)具有更高的轴向长宽比,其独特的分子特征也更易增加血液黏度[6]。国外多个病例报道提示,早期出现高黏滞血症的IgM型MM患者较高危,建议尽早进行血浆置换,并积极全身治疗,减少异常单克隆免疫球蛋白的产生,尽快达到疾病深度缓解[7,8,9]。
华氏巨球蛋白血症(Waldenstroms macroglobulinemia,WM)是一种淋巴浆细胞肿瘤,以分泌IgM型单克隆免疫球蛋白为特征,常表现为骨髓浸润、高黏滞血症、淋巴结肿大或肝脾大等,临床表现与IgM型MM有许多相似之处,难以鉴别。但溶骨性病变是MM特征性表现,在WM患者中罕见,有助于两者的区分。其次,WM患者大多有MYD88 L256P基因突变,而MM中常无这一突变[10]。此外,IgM型MM患者IgH易位阳性率极高,其中t(11;14)最为常见,可在40%患者中发现这一遗传学改变[6,11]。而WM患者最常见的细胞遗传学改变为6q-,几乎不伴IgH易位,因此检测IgH易位与6q-对鉴别WM与IgM型MM也有一定意义。
国外有研究回顾性分析17例IgM型MM患者,可在4例患者外周血中发现大量浆细胞,并有2例进展为浆细胞白血病,提示IgM型MM患者较常见亚型进展为浆细胞白血病的风险高[12]。本组患者中,有4例可在血涂片中发现浆细胞,虽然外周血浆细胞比例较低(均<5%),但不能否认IgM型MM髓外进展风险高,提示我们对IgM型MM患者应进行细致的血涂片检查或外周血流式细胞术检测,避免浆细胞白血病的漏诊。此外,4例外周血浆细胞阳性的患者多伴有t(11;14),与国外报道结果一致,提示t(11;14)可能是MM进展为浆细胞白血病的高危因素[12]。
在治疗方面,国外一项大型多中心研究纳入134例IgM型MM患者,发现这一亚型对传统化疗药物、蛋白酶体抑制剂等均较为敏感,且预后尚可,中位OS时间可达61个月,与常见MM亚型无明显区别[6]。既往国内有研究报道4例IgM型MM,提示这一罕见亚型进展迅速,预后相对较差,可能与新药应用较少有关[13]。本组6例患者均接受以蛋白酶体抑制剂等新药为基础的治疗,治疗敏感性较好,疗效均至少获得PR,与国外研究结果类似[6,12]。但本组患者病例数较少,且部分患者随访时间较短,无法准确评估IgM型MM患者的生存期。自体移植是MM系统治疗中的重要一环,但有研究表明对IgM型MM患者进行自体移植并不能改善预后,生存期相对较短[14]。此外,有研究提示Bcl-2抑制剂对携带t(11;14)重排的MM患者具有较好疗效[15],因此维奈托克治疗IgM型MM可能会改善其预后,并延长患者生存期。
IgM型MM非常罕见,具有一定的临床特点,除具有骨髓瘤典型临床表现外,也可伴高黏滞血症、淋巴结肿大或肝脾大等淋巴浆细胞肿瘤的特征表现。同时,t(11;14)易位是IgM型MM最常见的细胞遗传学异常。此外,在IgM型患者外周血中常能发现浆细胞,提示这一亚型髓外进展风险高。在治疗反应上,IgM型MM对目前常用治疗药物均较为敏感,疗效尚可。综合国外既往报道,与常见亚型相比,IgM型MM未显示明显预后不良。

























