
报告9例遗传性椭圆形红细胞增多症(hereditary elliptocytosis,HE)患者基因突变类型并分析HE致病性基因突变的特征。
报告中国医学科学院血液病医院2018年6月至2022年2月临床诊断的9例HE患者临床及基因突变特征,应用Sanger测序方法进行验证,分析基因突变构成、突变类型、基因型及与临床表型之间的关系。
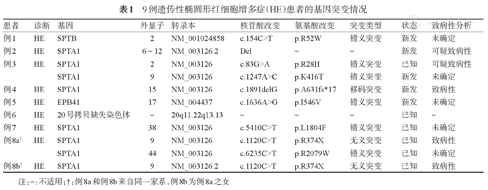
9例HE患者中6例为SPTA1、1例为SPTB、1例为EPB41红细胞膜蛋白基因突变,另1例为20号染色体拷贝缺失。共涉及11个基因突变位点,包括6个已知突变和5个新发突变。5个新发突变分别为:SPTA1基因9号外显子c.1247A>C(p.K416T),15号外显子c.1891delG(p.A631fs*17),6~12号外显子Del;SPTB基因c.154C>T(p.R52W);EPB41基因c.1636A>G(p.I546V)。6例SPTA1突变患者中3例为SPTA1 9号外显子突变。
SPTA1是HE患者最常见的突变基因。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
遗传性椭圆形红细胞增多症(hereditary elliptocytosis,HE)是以外周血椭圆形红细胞增多为特征的遗传性溶血性疾病。由编码红细胞膜或骨架蛋白的基因突变引起红细胞膜异常所致,主要呈常染色体显性遗传,主要涉及SPTA1、SPTB和EPB41基因,分别编码ɑ血影蛋白、β血影蛋白和带4.1蛋白[1]。HE在世界各地均有分布,在欧美国家患病率为0.03%~0.05%[2],在非洲等疟疾流行地区患病率可达0.6%~1.6%[3]。文献中HE多为散在的个例或家系报道,多数HE无明显临床症状,容易与遗传性球形红细胞增多症混淆。了解HE的基因突变特点,以及基因型与临床表型的关系对于HE的精准诊断具有重要的意义。我们报告本中心确诊的9例HE患者,总结HE基因突变特点如下。
本研究为回顾性研究,以2018年6月至2022年2月中国医学科学院血液病医院贫血诊疗中心连续收治的8个独立家系的9例HE患者为研究对象。患者经详细的病史采集、体格检查、实验室和影像学检查,并应用二代基因测序(NGS)确定基因型,依照文献[4]标准明确诊断为HE。
血常规,外周血涂片,游离血红蛋白(F-HB),结合珠蛋白(HP),成熟红细胞寿命测定(内源性CO呼气试验),红细胞膜病相关溶血试验[红细胞渗透脆性试验(EOF)、酸化甘油溶血试验(AGLT50)、伊红-5′-马来酰亚胺(EMA)检测],红细胞酶病相关溶血试验[葡萄糖-6-磷酸脱氢酶(G6PD)、丙酮酸激酶(PK)、红细胞嘧啶5’-核苷酸酶(P5’N)],血红蛋白病相关溶血试验[血红蛋白A2(HbA2)、血红蛋白F(HbF)],获得性溶血性贫血试验[直接抗球蛋白试验(Coombs)、酸溶血试验、冷凝集素试验],消化系统超声检查等。
参照文献[5]方法进行NGS检测,平均测序覆盖度为99.59%,平均测序深度569.67×,将测序结果与人类基因突变数据库(HGMD)(http://www.hgmd.cf.ac.uk/ac/index.php)及千人基因组数据库进行比对。根据美国医学遗传学与基因组学会(ACMG)发布的变异解读指南进行致病性分析,并结合患者的临床特点和家族史,对患者及其父母的DNA采用了Sanger测序法进行突变位点验证,从而确定患者的致病性突变。
9例患者中,男3例,女6例,中位年龄40(26,55)岁,中位确诊年龄为32(21,40)岁,中位病史8(0.5,20)年。8例脾脏肿大,5例皮肤及巩膜黄染,4例明显乏力,3例尿色加深,2例间断发热。有贫血相关家族史者6例。
9例患者中有8例存在轻中度贫血,中位HGB 84(74,93)g/L;中位WBC 5.31(2.81,5.44)×109/L,中位PLT 184(147,238)×109/L,中位网织红细胞比例(RET%)10.69%(7.85%,14.56%),中位网织红细胞绝对值(ARC)0.236(0.182,0.379)×109/L,中位平均红细胞体积(MCV)104.4(97.0,114.9)fl,中位平均血红蛋白浓度(MCH)34.7(32.5,36.8)pg,中位平均红细胞血红蛋白浓度(MCHC)333(320,342)g/L,中位红细胞体积分布宽度(RDW-SD)84.75(79.73,87.38)fl。7例检验了血清LDH的患者中5例高于正常值(>247 U/L)。5例患者的F-HB高于正常值上限(40 mg/L),6例患者HP低于正常值下限(0.5 g/L)。4例患者(例2、例5、例7、例8b)检测了红细胞寿命,分别为9、24、13、14 d。7例患者AGLT50试验阳性(<290 s)。在8例进行了EOF试验的患者中7例显示开始溶血时间延长,红细胞渗透脆性增加。6例患者EMA试验均正常。
外周血涂片可见成熟红细胞大小不一,椭圆形红细胞占50%~90%,偶见球形红细胞和红细胞碎片。骨髓涂片有核细胞增生明显活跃,红系比例明显增高(35.3%~77.5%)。
9例患者检出红细胞膜蛋白突变基因分别涉及SPTA1、SPTB和EPB41,共检出11个基因突变位点。其中SPTA1基因突变6例(分别为单等位基因杂合突变3例、双位点复合杂合突变2例、6~12号外显子片段缺失1例),SPTB突变1例,EPB41突变1例,20号染色体拷贝缺失1例。11个突变的基因位点包含9个基因点突变和2个基因大片段缺失,其中5个为新发突变,6个为已知突变。5个新发突变分别为:SPTA1基因的9号外显子c.1247A>C(p.K416T);15号外显子c.1891delG(p.A631fs*17)为致病性突变;6~12号外显子Del为可疑致病性;SPTB基因c.154C>T(p.R52W)和EPB41基因c.1636A>G(p.I546V),致病性分析均为未确定。6个已知突变中有5个来自SPTA1基因,另一个为20号染色体拷贝缺失(表1)。
9例遗传性椭圆形红细胞增多症(HE)患者的基因突变情况
9例遗传性椭圆形红细胞增多症(HE)患者的基因突变情况
| 患者 | 诊断 | 基因 | 外显子 | 转录本 | 核苷酸改变 | 氨基酸改变 | 突变类型 | 状态 | 致病性分析 |
|---|---|---|---|---|---|---|---|---|---|
| 例1 | HE | SPTB | 2 | NM_001024858 | c.154C>T | p.R52W | 错义突变 | 新发 | 未确定 |
| 例2 | HE | SPTA1 | 6~12 | NM_003126.2 | Del | - | - | 新发 | 可疑致病性 |
| 例3 | HE | SPTA1 | 2 | NM_003126 | c.83G>A | p.R28H | 错义突变 | 已知 | 可疑致病性 |
| SPTA1 | 9 | NM_003126 | c.1247A>C | p.K416T | 错义突变 | 新发 | 未确定 | ||
| 例4 | HE | SPTA1 | 15 | NM_003126 | c.1891delG | p.A631fs*17 | 移码突变 | 新发 | 致病性 |
| 例5 | HE | EPB41 | 17 | NM_004437 | c.1636A>G | p.I546V | 错义突变 | 新发 | 未确定 |
| 例6 | HE | 20号拷贝缺失染色体 | - | 20q11.22q13.13 | - | - | - | 已知 | - |
| 例7 | HE | SPTA1 | 38 | NM_003126 | c.5410C>T | p.L1804F | 错义突变 | 已知 | 未确定 |
| 例8a† | HE | SPTA1 | 9 | NM_003126 | c.1120C>T | p.R374X | 无义突变 | 已知 | 致病性 |
| SPTA1 | 44 | NM_003126 | c.6235C>T | p.R2079W | 错义突变 | 已知 | 未确定 | ||
| 例8b† | HE | SPTA1 | 9 | NM_003126.2 | c.1120C>T | p.R374X | 无义突变 | 已知 | 致病性 |
注:-:不适用;†:例8a和例8b来自同一家系,例8b为例8a之女
3例患者(例3、例8a、例8b)携带SPTA1基因9号外显子突变,贫血程度各不相同。例3、例8a均有SPTA1基因的复合杂合突变,例3呈中度贫血,例8a外周血涂片中可见椭圆形红细胞且伴有脾大症状,但无贫血。例8b与例8a来自同一家系,为例8a之女,仅遗传了父亲的SPTA1突变c.1120C>T (p.R374X),呈中度贫血。
有3例患者(例1、例3、例5)进行了父母亲代基因突变位点的Sanger测序验证。其中例1 SPTB基因2号外显子的突变c.154C>T(p.R52W)经验证源自父亲。例3 SPTA1基因复合杂合突变,c.83G>A(p.R28H)源自父亲,c.1247A>C(p.K416T)源自母亲。例5 EPB41基因17号外显子突变c.1636A>G(p.I546V)遗传自母亲。
HE常见突变的红细胞膜蛋白基因涉及SPTA1、EBP41和SPTB。SPTA1是最常突变的基因,且突变主要发生在2号外显子,以点突变为主,错义突变是最主要的突变类型。HE的基因型和临床表型间似无明显相关性。我们推测SPTA1基因2号外显子上的c.82-c.83区域突变所致的28号氨基酸改变,40号外显子的c.5572C>G(p.Leu1858Val)可能为HE患者SPTA1基因的高频突变。
为了探究HE基因型与临床表型之间的潜在关联,我们汇总了Pubmed(检索词:hereditary elliptocytosis)2000至2022年国内外进行了基因检测的HE病例并对其进行分析。来自不同家系的30例HE患者(文献报道22例,本研究8例)共涉及36个膜蛋白基因位点(附表)。其中,SPTA1(27/36,75.0%)是HE患者最常见的突变基因,其次分别是EPB41(7/36,19.4%)和SPTB(2/36,5.6%)。27例SPTA1突变的基因位点中,24例为点突变,且SPTA1突变主要发生在2号外显子(8/24,33.3%),同时也在其他外显子散在分布。在中国患者的14个SPTA1基因突变位点中,突变仍主要发生于2号外显子(4/14,28.6%)。错义突变(18/24,75%)是SPTA1最主要的点突变类型。无义突变(3/6,50%)是EPB41基因最主要的点突变类型。
综合文献报道与本研究病例,SPTA1基因2号外显子发生突变的8例HE患者贫血程度各不相同,这或许与突变位点的不完全外显有关,其中一个基因很可能发生沉默并未表达。Motulsky等[6]也曾有类似推测,可通过检测相应mRNA表达进一步验证。来自不同家系的3例HE患者均为SPTA1基因c.82C>T(p.R28C)突变[7,8,9],分别呈轻/中度贫血或不贫血。同一家系中有SPTA1基因c.1120C>T(p.R374X)致病突变的父亲(例8a)无贫血,女儿(例8b)呈中度贫血。由于不同个体间SPTA1基因编码α链数量是可变的,因此相同基因突变的不同个体间临床表现不尽相同。
SPTA1基因位于1q23.1上,包含52个外显子,其所编码的α血影蛋白是红细胞骨架蛋白的主要成分,该基因突变时将影响红细胞骨架的水平联结,导致红细胞的稳定性和变形能力下降发生溶血[10]。既往研究发现,α血影蛋白NH2末端区域的错义突变是HE和遗传性热异形红细胞增多症(HPP)中最常见的缺陷之一[11]。28号密码子突变所致氨基酸Arg28改变是非洲裔美国人SPTA1基因的热点突变[12]。依据本次文献汇总结果,3例不同家系患者中均发现了SPTA1基因2号外显子错义突变c.82C>T (p.R28C)[7,8,9];3例不同家系患者SPTA1基因的2号外显子错义突变c.83G>A(p.R28H)[7,13]。故推测28号氨基酸在α血影蛋白的结构和功能中发挥着重要的作用。在两个不同家系患者中发现SPTA1基因的40号外显子错义突变c.5572C>G(p.Leu1858Val)[7,14]。因此我们推测2号外显子上的c.82-c.83区域突变所致的28号氨基酸改变,40号外显子的c.5572C>G (p.Leu1858Val)可能为HE患者SPTA1基因的高频突变。
EBP41基因位于1p35.3上,包含28个外显子,编码4.1R蛋白,该基因突变所致的4.1R蛋白缺陷会影响细胞膜骨架完整性从而发生溶血。4.1R的部分缺乏可见于白种人HE患者,表现为轻度HE,很少或几乎没有溶血[15,16,17,18]。4.1R蛋白完全缺失患者会发生严重溶血[19,20,21,22]。故4.1R蛋白缺陷程度与患者的临床表型密切相关,单一杂合EPB41基因突变的个体有轻/中度贫血,而EPB41基因纯合突变所致4.1R蛋白完全缺失患者表现为重度贫血。
SPTB基因位于14q23.3上,包含38个外显子,编码的β血影蛋白是细胞膜骨架的重要组成部分。β血影蛋白COOH末端区域的截断突变(包括插入、缺失、无义突变等)会影响ɑ-β二聚体重复区进而影响四聚体结构的形成,这是HE或HPP常见致病缺陷[11,23]。结合文献报告发生SPTB突变的HE患者仅有2例:c.6203 T>C(p.L2068P)[24]和c.154C>T(p.R52W)均为错义突变,均为中度贫血。因病例数较少,故SPTB基因与HE临床表型之间关系仍是未知。
我们的研究也存在不足之处,由于大部分HE患者没有临床症状不易发现,因而目前报道的完善基因检查的样本例数较少,HE的基因型与表现型之间的对应关系尚未阐明。与例6相似,既往曾报道伴有染色体20q-的MDS患者外周血涂片发现椭圆形红细胞增多但原因尚不明[25,26,27]。
NGS技术对于遗传性疾病的诊断提供了有力支持,相信随着NGS的广泛应用和病例数的累积,HE基因型与表现型之间的关系将更加明晰,从而实现遗传性疾病的分子诊断新时代。
所有作者声明无利益冲突

























