
骨微结构的破坏是骨质疏松症最主要的病理性改变之一。骨微结构损伤会导致骨强度下降,骨折风险增加。骨重建失衡是骨质疏松症患者骨微结构损伤的主要原因。抗骨质疏松药物可以调节骨重建,从而改善骨微结构。目前抗骨质疏松药物主要包括抗骨吸收药、促骨形成药两大类。抗骨吸收药物[包括双膦酸盐、核因子κB受体活化因子配体(receptor activator of nuclear factor kappa-B ligand,RANKL)抑制剂等]一方面抑制骨吸收,防止骨小梁体积、数目及连接性的进一步损失;另一方面抑制骨重建,给骨重建单元提供更多时间来提高骨矿化程度。促骨形成药物(包括特立帕肽和阿巴洛肽)通过刺激骨重建及一定程度的骨塑建促使骨代谢正平衡(骨形成大于骨吸收);从而增加骨小梁数目,改善骨小梁结构。除以上两类药物之外,硬骨抑素抗体(如Romosozumab)可以中和硬骨抑素,具有双向作用,既可刺激骨形成,又可抑制骨吸收,对松质骨及皮质骨微结构的改善均有一定效果。既往有众多研究证实了各类抗骨质疏松药物在改善骨微结构、提升骨质量方面的疗效,但RANKL抑制剂促进骨塑建的具体作用机制、双膦酸盐对皮质骨孔隙度的影响等方面仍需要进一步研究。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
骨质疏松症是最常见的骨骼疾病。根据世界卫生组织(World Health Organization,WHO)的定义,骨质疏松症是一种以骨量减少、骨组织微结构破坏、骨脆性增加、骨折风险上升为特征的全身性骨骼疾病[1]。
骨质疏松症患者在日常生活中受到轻微外力或通常不会引起骨折的外力时,可能发生骨质疏松性骨折;常见于胸腰椎段椎体、髋部(股骨近端)、桡骨远端、肱骨近端以及骨盆和肋骨等部位[2]。我国关于骨质疏松性骨折(主要为髋部骨折和椎体骨折)的流行病学研究有限,但现有的不同地区的数据均显示中国大陆地区骨质疏松性骨折发生率正稳步增加[3]。此外,首次骨质疏松性骨折后1~2年内,患者再发骨折风险最高,再次骨折风险迫在眉睫[2]。
骨质疏松性骨折不同于普通的创伤性骨折,其发生在骨质疏松症病理性改变的基础上,是一种病理性骨折。骨微结构的破坏是骨质疏松症最主要的病理性改变之一[4,5]。骨微结构损伤会导致骨强度下降,骨折风险增加[6]。
松质骨的微结构改变主要发生在骨小梁,包括骨小梁逐渐丢失连接性、变得易于穿透、数量减少以及间隙增加,特别是以受力较小的水平支柱穿孔为主要特征[7]。一旦骨小梁连接性丢失,松质骨用于传导和承受应力的网状结构将受到破坏,机械性能随之下降[8]。皮质骨的微结构改变主要包括厚度变薄以及孔隙度增加[4],皮质内膜面逐渐发生小梁化病变[7,9],导致强度下降。
骨骼的生长代谢主要包括骨塑建与骨重建两个过程,与骨的生成、塑形、强度维持、骨量变化等息息相关。
骨骼在生理影响及机械应力作用下发生整体形状改变,导致骨骼逐渐调整到适应外力的状态,这个过程称为骨塑建。在骨塑建过程中,骨形成和骨吸收并不紧密耦联[10]。骨塑建界定骨的生成与发展、主导骨的形状与骨在空间中的移位,骨塑建异常可导致骨畸形或骨发育不良[10]。骨塑建也可见于老年人,基于骨塑建的骨形成有助于骨膜的扩增,正如基于重建的再吸收导致长骨和肋骨随年龄增长出现髓样扩张一样。骨塑建受遗传因素与环境因素的综合影响,如物理应力的刺激以及甲状旁腺素等激素的影响[11]。
骨重建是骨骼为了维持其强度和矿化稳态而进行的更新过程。骨重建包括持续清除散在旧骨中的骨单位,代之以新合成的蛋白基质,继而矿化形成新骨等过程。骨重建过程中,由不断重复、时空耦联的骨吸收和骨形成过程维持骨的完整性,并避免微损伤的积累[10]。成年前骨骼不断构建、塑形和重建,骨形成和骨吸收的正平衡(骨形成大于骨吸收)使骨量增加,并达到骨峰值;成年期骨重建达到平衡,维持骨量;此后随年龄增加,骨形成和骨吸收呈负平衡(骨形成小于骨吸收),骨重建失衡造成骨量丢失,过量的骨丢失导致骨质疏松症[12]。
在围绝经期和绝经后早期,雌激素水平下降,对破骨细胞的抑制作用减弱,导致骨吸收功能增强。尽管在此阶段女性骨形成的功能也有一定程度提高,但不足以代偿过度增加的骨吸收,骨吸收远大于骨形成的失衡状态造成骨微结构的破坏[12]。老年期骨质疏松症同样由于骨重建失衡导致进行性骨丢失,其特点是以增龄性成骨细胞功能降低为主,伴或不伴破骨细胞功能增强[12]。骨重建失衡,骨吸收/骨形成比例升高是骨微结构损伤的主要原因。皮质骨重建失衡会导致皮质骨孔隙度增加,骨量减少[10]。松质骨重建失衡会导致骨小梁穿孔、连续性中断,从而使骨小梁结构的连接性下降,从正常的板状结构转化为更薄的杆状结构,最终造成骨机械强度降低[13]。
抗骨吸收药物主要包括双膦酸盐、核因子κB受体活化因子配体(receptor activator of nuclear factor kappa-B ligand,RANKL)抑制剂、降钙素、雌激素及选择性雌激素受体调节剂类药物等。抗骨吸收药物作用机制各不相同(图1),但均可通过以下两方面改善骨微结构:一是抑制骨吸收,防止骨小梁体积、数目及连接性的进一步损失;二是抑制骨重建,给骨重建单元提供更多的时间来提高骨矿化程度[14]。因降钙素、雌激素及选择性雌激素受体调节剂类药物目前改善骨微结构和骨质量的证据有限,因此本文重点阐述双膦酸盐和RANKL抑制剂。
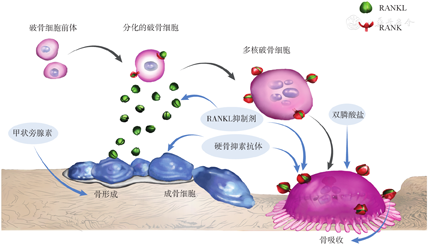
双膦酸盐的作用机制是抑制破骨细胞,从而降低骨转换,增加骨量和骨矿化程度[15];其效应来源于双膦酸盐被破骨细胞摄取,因此取决于双膦酸盐的累积剂量及其与骨的亲和力[16]。目前,双膦酸盐在改善松质骨及皮质骨的骨微结构和骨质量方面都有一定的研究证据。
骨小梁模式因子(trabecular bone pattern factor,Tb.Pf)是定量描述骨小梁连接性的一个指标。Tb.Pf值降低说明松质骨的构造由杆状向板状转变,因此Tb.Pf值越低表明骨连接性越好。在一项队列研究中,采用双膦酸盐治疗3年以上的骨质疏松性髋部骨折患者与未治疗的患者相比,Tb.Pf显著减低,骨连接性的保存更好,说明双膦酸盐可抑制骨吸收及有效保存骨量和骨质量[17]。一项在围绝经期女性中开展的Ⅲ期临床试验显示,阿仑膦酸钠治疗2~3年后,二维组织形态学检测发现骨小梁微结构改善,具体表现为骨体积增加、骨小梁厚度增加,以及骨小梁间隙减少;Micro-CT扫描显示,与安慰剂组相比,阿仑膦酸钠治疗组的骨体积更大、骨小梁数目更多。这一结果证实阿仑膦酸钠治疗可以维持和增加骨小梁厚度,减缓骨小梁丢失,减少单位骨小梁表面的骨重建点位数量,使现有的吸收部位得到更完整的填充,从而减少骨小梁局部变薄的部位而使其被动增厚[18]。阿仑膦酸钠的另一项Ⅲ期临床试验,在治疗第24个月或36个月时经髂骨获取绝经后骨质疏松症患者的骨活组织检查标本24例,发现阿仑膦酸钠治疗明显增加了骨质矿化程度及均一性,且皮质骨孔隙度显著下降[19]。然而在一项前瞻性非随机开放标签的研究中,采用唑来膦酸治疗绝经后骨质疏松症患者18个月,未观察到皮质骨孔隙度发生明显变化,而胫骨皮质厚度及骨小梁数量显著增加[20]。
尽管双膦酸盐治疗可以带来骨微结构的改善,但也存在长期应用后过度抑制骨重建,造成微损伤积累的问题。微损伤是指在某些高分辨率影像学检查中[如同步辐射X线显微断层扫描(synchrotron radiation X-ray microcomputed tomography,SR-μCT)]可见的穿孔或微骨折。其中穿孔是指骨小梁某个区域完全破坏,归咎于破骨细胞的作用;微骨折是指显微镜下可见的骨折,具有典型的线形锐利边缘,长度约30~100 μm[21]。有学者探讨了双膦酸盐治疗对骨小梁微结构的影响,包括穿孔和微骨折,发现与未接受双膦酸盐治疗的股骨颈骨折患者,以及未经任何抗骨质疏松药物治疗且未发生骨折的患者相比,接受阿仑膦酸钠长期治疗的骨折患者骨小梁微骨折总体积更多;但未接受双膦酸盐治疗的股骨颈骨折患者的骨小梁穿孔最为密集,体积也最大。该研究提示,可能有一部分患者不能从双膦酸盐治疗中获得保护效应,其微结构损伤和骨骼脆性增加,导致骨骼机械强度下降,从而增加了骨折风险[21]。还有学者认为,长期使用双膦酸盐治疗除引起微损伤积累外,还可能因长期抑制骨转换而导致骨重建单元有更多的时间二次矿化,从而导致过度矿化和同质性增加;尽管矿物质含量高的骨骼更加坚硬,但过度矿化和同质性增加会使骨骼变脆、易碎[22]。因此,对长期使用双膦酸盐治疗的患者,如能预测微骨折积累的关键时间点,则有可能优化其治疗时长[21]。
RANKL是调节破骨细胞功能的重要分子,可与破骨细胞表面的核因子κB受体活化因子(receptor activator of nuclear factor kappa B,RANK)结合,促进破骨前体细胞向成熟破骨细胞分化,以及促进成熟破骨细胞发挥功能。RANKL抑制剂可特异性与RANKL结合,从而抑制破骨细胞的分化、成熟、活化,以减少骨吸收,提高骨密度与骨强度。截至目前,国内外获批上市的RANKL抑制剂仅地舒单抗注射剂一种。
地舒单抗是一种抗RANKL的全人源单克隆抗体,是首个被推荐用于治疗骨质疏松症的单克隆抗体。RANKL是成熟破骨细胞形成、活化和发挥功能的关键,地舒单抗能快速结合RANKL,减少骨吸收、抑制骨重建[23]。地舒单抗在提高骨质量方面能发挥重要作用,通过抑制破骨细胞的募集和(或)活性抑制骨重建,使骨重建空间被重新填满,表现为早期骨量增加;破骨细胞被抑制后,旧骨的二次矿化增加,促进了骨量提升,尤其是在皮质骨含量高的部位。FREEDOM研究证实,地舒单抗能够抑制破骨细胞的形成及活化,减少皮质骨侵蚀程度及面积,减少皮质下骨丢失,从而提升骨质量、增加骨强度[24]。FREEDOM研究的一项子研究表明,地舒单抗治疗36个月可降低股骨近端皮质的孔隙度,使基质矿化量和皮质厚度增加,从而提高骨强度,有利于减少髋部和非椎体骨折[25]。骨小梁评分(trabecular bone score,TBS)是一种基于双能X线吸收(dual-energy X-ray absorptiometry,DXA)图像的灰阶结构指数,能有效评估骨的微结构、描述骨的质量。TBS可直接衡量骨微结构和力学特征,并预测绝经后女性骨质疏松症患者当前及未来骨折的风险。FREEDOM研究的DXA子研究结果表明,地舒单抗治疗12、24及36个月时,腰椎TBS较安慰剂组显著增加[26]。
在地舒单抗治疗的10年内,患者椎体及髋部骨密度均呈持续而稳定的增长,而双膦酸盐治疗3~5年后骨密度增长进入平台期。究其原因,可能有以下三个方面因素的影响:①地舒单抗抑制骨吸收作用强于双膦酸盐;②地舒单抗更容易进入皮质骨部位,能减少皮质骨孔隙度;③地舒单抗治疗后在皮质骨内膜、骨小梁表面等部位基于骨塑建的骨形成(modeling-based bone formation,MBBF)较高。关于第二点,双膦酸盐在骨质中的结合依赖于其与羟基磷灰石的亲和力,而皮质骨表面骨代谢活性较低,羟基磷灰石基质暴露较少,因此双膦酸盐在皮质骨的部位结合较少;地舒单抗可直接与血液、组织间的RANKL结合,无需骨基质暴露。因此有学者认为因地舒单抗能更充分地分布到皮质骨,相较于阿仑膦酸钠可以更快速、更显著地抑制血清Ⅰ型胶原C-末端肽交联(serum C-terminal telopeptide of type 1 collagen,S-CTX)和血清Ⅰ型原胶原N-端前肽(procollagen type 1 N-peptide,P1NP)等骨转换标志物,发挥更强的骨重建抑制效应[17]。这一机制或可解释地舒单抗治疗后皮质骨密度明显增加的原因[17]。关于第三点,地舒单抗在促进MBBF的潜力上为骨质疏松治疗提供了新思路。Ominsky等[27]对卵巢切除的食蟹猴实施了为期16个月的皮下注射地舒单抗(25 mg/kg或50 mg/kg,每月1次);第6个月至第16个月股骨近端出现持续的MBBF,而基于骨重建的骨形成(remodeling based bone formation,RBBF)显著减少。一项地舒单抗联合特立帕肽的临床研究间接证实地舒单抗具有维持骨塑建的能力,当所有骨转换标志物被抑制时地舒单抗仍可增加面积骨密度[28]。地舒单抗治疗过程中观察到的骨量增加可能源于骨塑建发生、骨重建受到抑制、骨吸收形成的凹陷得到填充、二次矿化增加等多种因素的共同影响[27]。一项开放标签的Ⅳ期临床研究证实,地舒单抗增加了MBBF而减少了RBBF,在最大程度抑制骨重建的同时保留了骨塑建,在骨小梁与皮质骨内膜等容易发生骨吸收的部位形成新骨,从而实现长期治疗骨质疏松、持续提升骨密度的作用[29]。这表明与其他抗骨吸收药物相比,地舒单抗还显示出了一定的骨形成效应,MBBF可能是地舒单抗可长期治疗骨质疏松并提升骨密度的根源,但其具体作用机制仍不清楚。有学者推测地舒单抗的骨形成作用可能与成骨细胞中的RANKL反向信号传导相关[30]。最近,有研究证实了RANKL信号传导在成骨细胞生成中的作用以及RANKL反向信号传导在骨吸收和骨形成耦联中促进成骨细胞分化的作用[31,32]。因此,抑制骨髓间质干细胞中RANKL信号传导可能是地舒单抗引起超预期骨形成的原因。
骨矿化异质性对骨强度的作用以及是否存在理想的骨矿化异质性都是具有争议性的问题。有研究证实骨矿化异质性一方面可能是促使骨折裂纹发生的启动因素,另一方面也可阻止骨折裂纹的扩展[33]。理论上说,在长期抑制骨转换的情况下矿化程度(degree of mineralization of bone,DMB)可能会持续增加,而矿化异质性指数(heterogeneity index,HI)会持续降低,骨的材料属性可能会遭到破坏。为验证这一疑问,有学者纳入了FREEDOM研究中的92例及其延长研究中的46例女性患者,通过髂骨活检观察骨组织学、组织形态计量学和基质矿化特征。结果发现采用地舒单抗治疗10年后的骨组织形态正常,骨重建减少;治疗2年及3年时与安慰剂组相比,DMB增加、HI减低,但治疗5年后HI再无明显改变[34]。这些数据提示地舒单抗持续、强效的骨重建抑制不影响骨强度,并与其应用10年持续降低骨折风险的表现一致[35]。
目前的研究显示,地舒单抗停药后如果不进行后续序贯治疗,则可出现骨吸收增加、骨密度回落至治疗前水平,导致多发椎体骨折风险增加[36,37]。对地舒单抗停药的患者进行骨活检发现,皮质骨出现厚度减小的趋势,松质骨中骨小梁数目虽未减少但小梁厚度发生轻微改变。这在一定程度上解释了地舒单抗停药后临床椎体骨折风险增加到与安慰剂组水平相似的原因[38]。因此,对正在接受地舒单抗治疗的患者不应随意停药;若必须停用则建议采用其他抗骨吸收药物治疗(如双膦酸盐),以减缓骨密度下降及骨折风险反弹,并密切监测骨转换标志物和骨密度的变化[39]。
促骨形成药物主要包括甲状旁腺素类似物特立帕肽及阿巴洛肽。此类药物通过刺激骨重建及一定程度的骨塑建,促使骨代谢正平衡[12],作用机制如图1所示。
特立帕肽是重组人甲状旁腺素氨基端1~34活性片段(recombinant human parathyroid hormone 1~34,rhPTH 1~34)。目前已有多项研究证实特立帕肽在改善骨微结构及骨质量方面具有一定的作用。在骨质疏松症患者中,松质骨结构的破坏特征为板状结构变成杆状结构。结构模型指数(structure model index,SMI)是针对这一结构特征的量化指标,反映板状结构和杆状结构的比例。如果结构中骨小梁主要为板状结构,则SMI接近于0;反之,如果主要是杆状骨小梁,SMI则接近3。当骨小梁结构从板状向杆状转变时,该数值增大,反之亦然。从绝经后骨质疏松症患者中获取髂骨活检标本进行2D组织形态学检测后发现,特立帕肽平均治疗18个月后骨小梁及骨皮质厚度增加;与安慰剂组相比,特立帕肽治疗组松质骨的体积明显增加[40]。3D微计算机断层扫描显示,与治疗前相比特立帕肽治疗后骨小梁形态结构明显改善(更趋于板状结构,SMI显著降低),同时骨皮质厚度也有所增加[40]。研究还进一步发现,特立帕肽治疗12和18个月后腰椎及股骨颈骨密度的提升与治疗22个月时骨小梁微结构的改善(包括骨体积、骨小梁厚度、骨小梁分离度及SMI)呈显著正相关[40]。特立帕肽对椎体骨微结构的改善在绝经后骨质疏松症患者的研究中同样得到验证。Gallacher和Dixon[15]采用高分辨率CT扫描观察到特立帕肽治疗6个月后椎体骨密度及骨小梁骨微结构均得到改善,治疗12个月后骨小梁体积增加了30%,数量增加了19%。
股骨颈屈曲比(buckling ratio,BR)是皮质骨半径与厚度的比值,用于衡量皮质骨弹性不稳定性。BR增加提示骨皮质变薄,皮质骨的弹性不稳定性增加。Borggrefe等[41]研究证实,绝经后骨质疏松症患者经特立帕肽治疗24个月后,骨皮质厚度显著增加,BR显著下降,提示骨骼弯曲强度增加,骨骼稳定性改善。然而另一项经过18个月的研究显示,特立帕肽每天一次注射会导致桡骨和胫骨远端皮质骨孔隙度增加及骨密度下降[42]。尽管特立帕肽治疗对皮质骨微结构会产生一定的影响,但依旧能维持骨强度,这与特立帕肽的作用机制有关。特立帕肽提高骨转换水平,导致新形成的吸收腔明显增加,因此可以检测出孔隙度增加;同时特立帕肽提高骨形成的程度强于骨吸收,成骨细胞将分泌富含胶原的类骨质填充这些新形成的吸收腔,而类骨质的矿化需要一定的时间,因此骨矿物质密度发生暂时下降。但特立帕肽治疗对骨微结构的影响具有时限性,短期内对皮质骨微结构的不利因素可能并不影响长期、整体骨强度的维持。需要注意的是,特立帕肽终身累计使用时间不能超过24个月[43,44]。但在2002年于美国(2011年在中国大陆)上市后,长达15年的上市后监测结果提示特立帕肽并未增加骨肉瘤发生的风险[45]。2020年11月,美国食品药品监督管理局(Food and Drug Administration,FDA)取消终身累计不超过24个月的治疗限制与警告[46],期待未来能有研究深入探索特立帕肽更长治疗时间的可能性与临床益处。
特立帕肽与地舒单抗联合使用可避免单独应用特立帕肽诱导破骨细胞活化的不足,有效提升骨密度,改善骨微结构。一项为期15个月的开放标签随机对照研究纳入了绝经后重度骨质疏松患者76例,随机给予特立帕肽标准剂量(20 μg/d)联合地舒单抗或高剂量(40 μg/d)联合地舒单抗治疗,治疗15个月时与基线相比两组患者胫骨皮质组织矿物质密度和骨皮质厚度均显著增加[47]。
阿巴洛肽是一种甲状旁腺素相关肽衍生多肽类似物,是PTH1受体信号通路的选择性激活剂。雌性小鼠的动物研究结果显示,阿巴洛肽能增加股骨远端骨小梁体积分数、骨小梁连接性和数量,降低SMI的同时增加骨小梁厚度与骨皮质厚度[48]。在一项Ⅱ期临床研究中,222例绝经后骨质疏松症患者被分为五组,其中三组分别接受三种剂量(20、40、80 μg)的阿巴洛肽皮下注射、另两组分别接受特立帕肽和安慰剂治疗,治疗后采用TBS评估骨微结构的改善情况。结果发现根据年龄、体质指数及TBS值校正后,阿巴洛肽皮下注射各治疗组及特立帕肽治疗组TBS值相对安慰剂组显著增高,且阿巴洛肽80 μg皮下注射组又显著高于特立帕肽组[49]。阿巴洛肽上市时间较短,临床数据有限,其对骨微结构的作用还需要更多临床研究数据来佐证。目前,阿巴洛肽在中国尚未上市,缺少中国人群的临床数据。
双向作用药物主要是硬骨抑素抗体(sclerostin-antibodies,Scl-Ab),可中和硬骨抑素,从而刺激骨形成并抑制骨吸收,作用机制见图1。
目前研究表明,Scl-Ab治疗可显著增加骨小梁、骨膜、皮质内膜和皮质内部表面的骨形成,增加骨量,提升骨强度[50]。Romosozumab是一种中和硬骨抑素的人源性单克隆抗体,已获FDA批准用于治疗高骨折风险的绝经后骨质疏松症患者[51,52]。动物研究提示,Romosozumab可增加面积骨密度、体积骨密度、骨小梁和皮质骨质量以及骨强度[53];并且骨小梁、骨膜、皮质内膜和皮质内部表面的骨形成与Romosozumab呈剂量依赖性增加[54]。对Romosozumab作用机制的进一步研究发现,Scl-Ab治疗早期骨量增加、皮质和骨小梁形成增加与破骨细胞活性下降有关,后续增加则源于残留的皮质内与骨小梁内成骨细胞刺激以及持续较低的破骨细胞活性[55]。此外,与Scl-Ab相关的新骨形成多源于MBBF[56]。随后多项研究证实,Romosozumab治疗能改善骨微结构并提高骨质量。一项Ⅰb期随机双盲对照研究结果提示,Romosozumab相对于安慰剂治疗能显著降低骨小梁分离度,增加骨小梁骨密度、体积分数以及皮质厚度,且在3个月的随访期间得到持续改善[57]。一项国际性随机双盲安慰剂对照的平行研究结果表明,Romosozumab治疗可带来早期短时的骨形成增加,并持续减少骨吸收,抗吸收效应最终导致骨转换减低。这种综合的作用机制可以显著增加骨量,改善骨小梁厚度和连接性[58]。
需要注意的是,Romosozumab的一项Ⅲ期临床研究数据显示,与阿仑膦酸钠相比Romosozumab治疗与更多的心血管事件相关,需进一步关注。Romosozumab的临床研究数据支持连续使用12个月,更长时间的临床疗效还没有证据。目前Romosozumab的注册临床试验在我国正在开展。
综上所述,骨质疏松症是一种以骨强度下降和骨折风险上升为特征的全身性骨骼疾病。骨强度可以从骨密度与骨质量两个维度来评价。在诸多因素中,骨微结构是决定骨质量的关键因素。骨重建失衡引起的骨吸收/骨形成比值增加是导致骨微结构损伤的主要原因。目前,抗骨质疏松药物主要包括抗骨吸收药物和促进骨形成药物两大类。抗吸收药物双膦酸盐具有抑制破骨细胞的作用,可降低骨转换,增加骨量、骨矿化程度,因此可以增加骨小梁厚度,减缓骨小梁丢失;双膦酸盐在降低皮质骨孔隙度方面存在争议,并且长期治疗后会过度抑制骨重建,造成微损伤积累,一定程度上影响骨强度,需注意治疗时长。抗吸收药物地舒单抗可快速结合RANKL,抑制破骨细胞形成及其功能,从而减少骨吸收,抑制骨重建。地舒单抗同时作用于皮质骨和松质骨,增加骨皮质厚度、降低骨皮质孔隙度、改善TBS;同时增加MBBF,减少RBBF,长期治疗可持续提升骨密度,持续、强效地抑制骨重建而不影响骨强度。促骨形成药物特立帕肽及阿巴洛肽通过刺激骨重建及一定程度的骨塑建促使骨代谢正平衡,从而增加骨小梁体积和数目、改善骨小梁形态结构、增加骨皮质厚度,增加骨骼的弯曲强度;可能对皮质骨产生一定不良影响,包括增加孔隙度、降低皮质骨密度,但不影响整体骨强度。新型的双向作用药物硬骨抑素抗体中和硬骨抑素刺激骨形成和抑制骨吸收,降低骨小梁分离度,增加骨小梁骨密度、体积分数以及皮质厚度等。
目前已有相当一部分研究证据证实了各类抗骨质疏松药物在改善骨微结构、提升骨质量方面的疗效。但某些药物的作用机制或效果仍需要进一步探讨,如RANKL抑制剂增加MBBF、减少RBBF的机制及双膦酸盐对皮质骨孔隙度的改善等。新型药物Scl-Ab可显著增加骨量,改善骨小梁厚度和连接性,目前结果令人鼓舞,其应用前景值得期待。
本研究得到安进公司医学部李宗杰、朱志伟、李青芳、李小慧对文献检索的支持和帮助

























