
基于二氧四氢喋啶合酶(LS)多聚纳米抗体,建立一种检测血清中可溶性细胞程序性死亡-配体1(PD-L1)蛋白的方法。
采用竞争ELISA筛选识别PD-L1不同表位的纳米抗体作为配对抗体,建立双纳米抗体夹心ELISA检测方法(Nbs-ELISA)。为了进一步提高敏感性,将纳米抗体与LS进行融合,获得多聚形式的纳米抗体作为sPD-L1蛋白的捕获分子,建立基于LS的多聚纳米抗体ELISA检测方法(LSNbs-ELISA)。
与Nbs-ELISA方法相比,LSNbs-ELISA方法对sPD-L1的检测敏感性提高了约11倍,Nbs-ELISA对血清中sPD-L1的检出限为2.87 ng/ml,而LSNbs-ELISA对PD-L1的检出限为0.255 ng/ml。2种检测方法对sPD-L1的检测均具有很高的特异性,与掺入血液中的PD-1、CD27、CD70、CD137和CD147蛋白均不结合。
建立的基于LS多聚纳米抗体检测血清中PD-L1的检测方法有较高的敏感性和特异性,对肺癌患者血清中可溶性PD-L1的检测具有潜在的临床应用价值。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
T细胞膜上的程序性死亡受体-1(programmed death-1,PD-1)与位于抗原提呈细胞膜上的配体PD-L1结合,介导负调控信号通路,抑制T细胞的增殖和功能[1]。肿瘤细胞通过表达PD-L1,抑制细胞毒性T淋巴细胞的增殖和功能,参与肿瘤的免疫逃逸。近年来,针对免疫检验点分子PD-1和PD-L1的免疫检验点阻断(immune checkpoint blockade,ICB)疗法对多种实体瘤显示出显著的临床疗效。基于ICB的免疫治疗正在成为包括非小细胞肺癌(non-small cell lung cancer,NSCLC)在内的多种实体瘤的有效临床治疗手段。
可溶性免疫检查点(soluble immune checkpoint,sICP)分子是近年来发现的存在于多种肿瘤患者血浆中的生物标志物[2,3,4]。PD-L1存在膜型(mPD-L1)和可溶性(sPD-L1)2种,mPD-L1胞外段可被基质金属蛋白酶裂解从而脱落形成sPD-L1。研究结果表明,高水平的sPD-L1可能与肺癌的不良预后相关,并与癌症病程和免疫治疗疗效相关[5],肺癌患者外周血中sPD-L1含量明显高于健康人[6]。然而,这些免疫调节蛋白在外周血中的含量普遍较低,如健康人血浆中sPD-L1的质量浓度为0.75~1.04 ng/ml[7]。目前,用于检测血清蛋白标志物的酶联免疫吸附测定(enzyme linked immunosorbent assay,ELISA)方法的检测下限为1~2 ng/ml,其敏感性不足,因此需要开发新的更加敏感的检测方法。
纳米抗体是源自骆驼科动物重链抗体可变区的单域抗体,具有相对分子质量(15 000)小、制备简便经济、易修饰和结构改造、性质稳定、耐受极端环境等特点[8,9,10,11]。这使纳米抗体在分子检测领域具有无可比拟的优势。本研究中,采用结合双表位的PD-L1纳米抗体建立了夹心ELISA检测sPD-L1的方法,其敏感性与采用单抗的ELISA方法相当。为了进一步提高检测的敏感性,还采用多聚形式的纳米抗体以获得更高的检测效果。二氧四氢喋啶合酶(lumazine synthase,LS)是源自超嗜热菌Aquifex aeolicus的一种能自组装形成多聚形式的生物纳米材料,在疫苗研究中被广泛作为多肽疫苗的展示平台[12,13,14]。LS能够自组装成内径为9 nm、外径为15 nm左右的二十面体纳米颗粒,并可在大肠杆菌中表达,是一种较为理想的多聚化展示平台。因此,本研究中将PD-L1纳米抗体与LS进行融合,利用其自组装效应使纳米抗体形成多聚形式,以期提高捕获抗原的能力,并期望开发一种成本低廉、灵敏度较高的双纳米抗体夹心ELISA检测血液中sPD-L1的方法,为今后评估肺癌患者的病程和免疫治疗效果提供一种新的方法。
人PD-L1、PD-1、CD27、CD70、CD137和CD147重组蛋白(北京义翘神州科技有限公司),3,3′,5,5′-四甲基联苯胺(tetramethyl benzidine,TMB,上海源叶生物科技有限公司),辣根过氧化物酶(horseradish peroxidase,HRP)标记的抗血凝素(hemagglutinin,HA)标签小鼠单克隆抗体(北京义翘神州科技有限公司),Nco Ⅰ、Not Ⅰ限制性内切酶、T4连接酶[宝日生物工程(大连)有限公司],Ni Sepharose 6 Fast Flow(美国GE公司),96孔酶标板(美国Corning有限公司)。PD-L1特异性纳米抗体P3F11、F2H9、P3C8和F3A6由本研究组前期通过噬菌体展示技术筛选获得。
挑取单个BL21(DE3)重组菌,接种至2×YT液体培养基,震荡培养直至600 nm下的吸光度值(A值)为0.5~0.6;加入终浓度为0.2 mol/L的异丙基-β-D-硫代半乳糖苷(isopropyl-beta-D-thiogalactoside,IPTG),在30 ℃,180 r/min震荡条件下培养12 h;8 000 r/min离心15 min后,收集诱导后的菌体,用磷酸盐缓冲液(phosphate buffer saline,PBS)重新悬浮,并重复2次,超声破碎,12 000 r/min离心10 min收集上清,采用Ni亲和柱纯化、500 mmol/L咪唑洗脱所需蛋白,十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulphate-polyacrylamide gel electrophoresis,SDS-PAGE)分析纯化蛋白,BCA(bicinchoninic acid)蛋白定量试剂盒对纳米抗体纯化蛋白进行定量。
在UniProtKB数据库中检索源自超嗜热菌的LS蛋白O66529序列,在金唯智公司(苏州)经过反向翻译和大肠杆菌密码子优化后,合成含有5′ Not I和3′ Xho I的DNA序列,并将其克隆到pET22b表达载体。纳米抗体基因用限制性内切酶Nco Ⅰ和Not Ⅰ从pMECS载体上切下后,连接到构建的pET22b-LS中,纳米抗体和LS之间采用GSGGSGGSG连接。连接产物转化大肠杆菌感受态细胞BL21(DE3)。按照纳米抗体的表达纯化方法制备融合了LS的纳米抗体蛋白。
用pH值为9.6的碳酸盐缓冲液将PD-L1重组蛋白稀释至1 μg/ml,后包被96孔酶标板,4 ℃静置过夜;经0.05% PBST洗后,用5%脱脂奶粉在37 ℃封闭2 h后加入梯度稀释的100 μl纳米抗体,37 ℃孵育1.5 h;然后加入1∶4 000稀释的HA-HRP,37 ℃静置孵育1 h;最后加入100 μl 3,3′,5,5′-四甲基联苯胺(3,3′,5,5′-tetramethylbenzidine,TMB)显色液避光显色10 min后立刻加入50 μl 2 mol/L的H2SO4终止反应。酶标仪上测定各孔在450 nm下的吸光度值(A450值)。各实验组均设置3个重复孔。
采用竞争ELISA法分析配对纳米抗体(以VHH-1和VHH-2表示)之间是否结合共同表位。将100 μl用包被液稀释至1 μg/ml PD-L1重组抗原加入96孔酶标板4 ℃包被过夜,经0.05% PBST洗涤和5%脱脂奶粉37 ℃封闭2 h后,加入不同浓度含HA标签的结合抗体VHH-1,37 ℃孵育1.5 h后,加入与VHH-1浓度相同的含c-Myc的竞争抗体VHH-2,PBST洗涤后,加入HRP偶联的鼠抗HA抗体37 ℃静置孵育1 h,后续显色和测定,按上述ELISA步骤操作,测定A450。竞争抗体VHH-2对结合抗体VHH-1的竞争率计算公式为
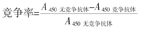
当竞争率≥50%时,两抗体结合同一表位;当竞争率<50%时,表明两抗体结合不同表位[15]。
将捕获纳米抗体包被于96孔酶标板孔中4 ℃静置过夜;经0.05% PBST洗净后,用5%脱脂奶粉37 ℃静置封闭2 h;加入梯度稀释后的抗原,37 ℃静置孵育1.5 h;加入检测纳米抗体37 ℃静置孵育1.5 h;加入HRP标记的抗检测纳米抗体载体所带标签的抗体37 ℃静置孵育1 h;TMB避光显色10 min后向孔中加入2 mol/L的H2SO4终止反应。利用酶标仪测定A450,计算P/N值并绘制热图,并依据A450抗原检测(P值)/A450阴性对照(N值)≥2.1为检测结果阳性的判定标准。
为了确定双纳米抗体夹心ELISA检测方法(Nbs-ELISA)、多聚纳米抗体ELISA检测方法(LSNbs-ELISA)中纳米抗体的最佳工作浓度,首先采用棋盘滴定法优化捕获纳米抗体和检测纳米抗体的工作浓度,根据不同抗原稀释浓度所得到的A450值计算P/N值。
通过检测双纳米抗体夹心ELISA法的检出限(limit of detection,LOD)确定其灵敏度。将PD-L1倍比稀释至不同浓度,以各蛋白稀释浓度为横坐标,检测所得的A450为纵坐标,绘制标准曲线。重复检测25份空白样品,并依据线性回归方程计算对应浓度计算LOD值,公式为
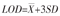
式中: 为浓度平均值;SD为浓度标准差。
为浓度平均值;SD为浓度标准差。
通过添加回收试验对血清中PD-L1蛋白进行检测,以评估Nbs-ELISA及LSNbs-ELISA方法的准确性[16]。分别配制不同PD-L1添加浓度的血清样品,利用Nbs-ELISA方法及LSNbs-ELISA方法进行5次重复检测。计算各添加浓度的平均回收率。
为了分析前期获得的4株PD-L1特异的纳米抗体P3F11、F2H9、P3C8和F3A6是否能够应用于夹心ELISA检测,需要制备含不同标签的PD-L1纳米抗体。将上述抗体基因分别构建到含有HA和c-Myc标签的pET22b表达载体中,转化BL21(DE3)表达菌并进行IPTG诱导表达。经Ni亲和柱纯化后,获得了较高纯度的重组纳米抗体,在SDS-PAGE胶19 000和22 000处分别显示带HA和c-Myc标签的纳米抗体条带,与预期蛋白大小一致(图1A)。为了确定携带不同标签的纳米抗体与PD-L1抗原的结合情况,利用间接ELISA法进行了检测,携带不同标签的纳米抗体均可以与PD-L1抗原结合,且呈浓度相关性(图1B),表明所带标签类型不影响纳米抗体与抗原的结合能力。
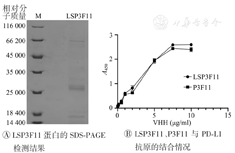
如图2A所示,经SDS-PAGE验证,纯化后的LSP3F11蛋白在18 000、27 000、66 200处均有清晰条带,可能为不同聚合形式的纳米抗体。以P3F11为对照,利用间接ELISA法检测LSP3F11与PD-L1抗原的结合能力(图2B),发现偶联了LS的P3F11对PD-L1抗原的结合与未偶联相比没有显著提高。
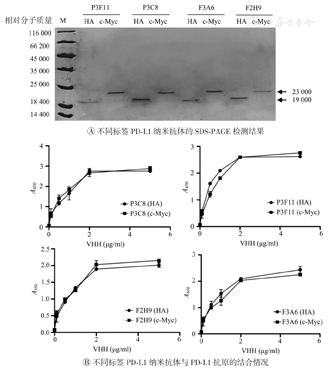
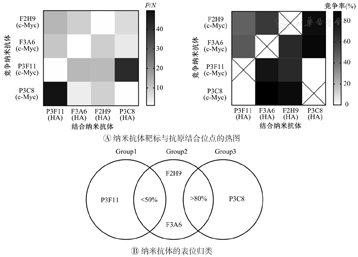
根据竞争率和特异性选择适用于夹心ELISA的抗体对。图3A所示为纳米抗体靶标与抗原结合位点的热图,色块深浅程度与纳米抗体之间靶向抗原结合位点竞争强度相反。如图3A,纳米抗体P3F11与P3C8靶标PD-L1结合表位的竞争率为0,因此其可用作夹心ELISA中的配对抗体。F2H9和F3A6对P3F11有弱的竞争作用(<50%),提示F2H9和F3A6与P3F11结合不同的表位,但位置较近。而F2H9和F3A6对P3C8有强竞争作用,竞争率分别为84%、89%,表明它们和P3C8结合的表位有重叠。因此,可以把这4种抗体按识别表位的不同,分成3组,其中P3C8和P3F11结合PD-L1上的2个独立表位,而F2H9和F3A6结合的表位与P3C8和P3F11都有交叉(图3B)。
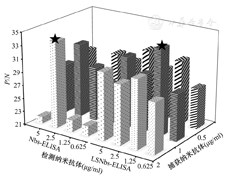
如图4所示,在Nbs-ELISA法中,当P3F11稀释至2 μg/ml,P3C8稀释至2.5 μg/ml时,可得最大P/N值。因此,确定捕获纳米抗体P3F11的工作浓度2 μg/ml,检测纳米抗体P3C8的工作浓度1.25 μg/ml为Nbs-ELISA的最佳工作浓度组合。在LSNbs-ELISA法中,当LSP3F11稀释至1 μg/ml,P3C8稀释至1.25 μg/ml时,可得最大P/N值。因此,确定捕获纳米抗体LSP3F11的工作浓度1 μg/ml,检测纳米抗体P3C8的工作浓度1.25 μg/ml为LSNbs-ELISA的最佳工作浓度组合。
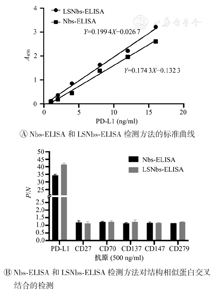
如图5A所示,Nbs-ELISA法的检出限为2.87 ng/ml,LSNbs-ELISA法的检出限为0.255 ng/ml。与P3F11相比,以LSP3F11融合蛋白作检测纳米抗体时,夹心ELISA检测方法的灵敏度提高了约11倍。如图5B所示,各实验组P/N值均<2.1,表明Nbs-ELISA和LSNbs-ELISA方法均与其他跨膜蛋白无交叉反应,具有较强的特异性。
如表1所示,Nbs-ELISA方法的回收率为82%~95%;LSNbs-ELISA方法的回收率为86%~95%。以《中国药典》2020年版重复性回收率限度为判定标准,表明2种检测方法在血清中检测的准确度较高。
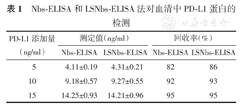
Nbs-ELISA和LSNbs-ELISA法对血清中PD-L1蛋白的检测
Nbs-ELISA和LSNbs-ELISA法对血清中PD-L1蛋白的检测
| PD-L1添加量(ng/ml) | 测定值(ng/ml) | 回收率(%) | ||
|---|---|---|---|---|
| Nbs-ELISA | LSNbs-ELISA | Nbs-ELISA | LSNbs-ELISA | |
| 5 | 4.11±0.19 | 4.31±0.21 | 82 | 86 |
| 10 | 9.18±0.57 | 9.27±0.55 | 92 | 93 |
| 15 | 14.25±0.93 | 14.21±0.96 | 95 | 95 |
本研究中,利用结合不同抗原表位的纳米抗体建立了用于检测sPD-L1的双纳米抗体夹心ELISA检测方法(Nbs-ELISA),该方法特异性良好,能准确检测出缓冲液中的PD-L1重组蛋白。此外,还利用可自组装的LS融合纳米抗体片段获得多聚纳米抗体,建立了基于多聚纳米抗体ELISA检测方法(LSNbs-ELISA),与其他报告结果相似,LS的存在未影响检测方法的特异性[17],且LSNbs-ELISA法较Nbs-ELISA法的LOD提高了约11倍,低至0.255 ng/ml,远低于健康人血浆中sPD-L1的质量浓度(0.75~1.04 ng/ml)。
目前,针对免疫检验点分子PD-1和PD-L1的免疫检查点抑制剂(immune checkpoint inhibitors,ICI)疗法对多种实体瘤患者,尤其是NSCLS患者显示出显著的临床疗效,但ICI单独使用时,针对NSCLC患者抗肿瘤的应答率只有20%~30%,大部分患者依然不能从ICI免疫治疗中获益[18]。采用液体活检可以对体液中的可溶性蛋白、核酸分子和循环肿瘤细胞等进行检测和筛查,具有便捷、成本低、患者接受度好等优势。纳米抗体因具有相对分子质量(15 000)小、互补决定区3(complementarity determining region 3,CDR3)较长、特异性高、易于改造等独特性质,在药物开发及检测方面中具有巨大的潜力[19]。在疾病诊断与治疗方面,Melli等[20]利用生物素化的纳米抗体建立了检测食源性致病菌志贺毒素的方法。Kandalaft等[21]的研究表明,高亲和力抗艰难梭状芽孢杆菌表层蛋白的纳米抗体能够抑制艰难梭状芽孢杆菌的运动并达到中和毒力因子毒性的目的。已有研究者利用纳米抗体建立了检测鸡血清中禽流感抗体的竞争ELISA方法[22];另有研究者利用纳米抗体碱性磷酸酶融合蛋白,建立了检测葡萄球菌肠毒素B的夹心化学发光免疫分析方法,其检测下限可达1.44 ng/ml[23]。根据Cortellis的数据显示,截至2022年1月,全球范围内纳米抗体药物的临床在研数量为31个。
LS是一种参与核黄素(维生素B2)生物合成的酶,由同源低聚物组成,其大小和亚基数量因其来源种属而异。由于其低聚性质,LS已被广泛用于多种生物医学领域,包括抗原呈递、药物递送等。来源于布鲁氏菌属的十聚体LS(BspLS)是第1个作为疫苗预防系统的LS。Mejias等[24]以BspLS为稳定支架,展示志贺毒素2型的B亚基,作为制备抗肠出血性大肠埃希菌抗体的策略。在药物递送中,LS被改造设计为一种药物递送载体,通过包封核酸实现RNA递送的改善[25]。二十面体的LS也被开发用在治疗癌症的靶向递送系统中。有研究将癌细胞靶向肽(如肿瘤脉管系统靶向肽)与AaLS基因融合,并连接抗癌药物(阿霉素或硼替佐米),发现采用了AaLS的靶向递送系统可与癌细胞特异性结合,并显著增强阿霉素和硼替佐米的细胞毒性作用[26]。另有研究者通过将IgG Fc结合结构域基因插入AaLS的C端,开发了多价抗体结合LS系统。由此得到的嵌合蛋白暴露出了Fc结合结构域,可以作为结合IgG分子进行靶向IgG递送的平台。该研究还表明,药物可以被包裹在AaLS系统的中空腔中,也可以化学偶联到AaLS的游离位点上[27]。
尽管目前LS在抗原呈递、药物递送系统中已有了较为广泛的应用,但其在分子检测中仍少有应用。本研究中,利用融合了LS的纳米抗体开发的LSNbs-ELISA法为未来检测肿瘤患者体液中的sPD-L1提供了基础。尽管LSNbs-ELISA法的灵敏度很高,但LSNbs的多聚形式尚不清楚,仍有待进一步研究。
所有作者均声明不存在利益冲突

























