
检测葡萄糖转运蛋白-2(GLUT-2)、葡萄糖激酶(GCK)在间歇低氧大鼠模型肝细胞中表达的变化,探讨间歇低氧引起胰岛素抵抗的相关机制。
24只6周龄健康雄性Sprague-Dawley(SD)大鼠按照随机数字表法分为对照组、间歇低氧4周组(IH4组)和间歇低氧8周组(IH8组),每组8只。间歇低氧组按预设通气模式每天给予间歇低氧暴露8 h,对照组给予间歇压缩空气,暴露时间同IH4组。实验结束后测定各组大鼠空腹血糖、空腹胰岛素,计算稳态模型评估-胰岛素抵抗指数(HOMA-IR)及胰岛素敏感性指数(ISI)。免疫组织化学染色观察肝细胞GLUT-2、GCK蛋白表达变化,并利用平均灰度值对蛋白进行定量分析。
与对照组相比,IH4组及IH8组空腹血糖、空腹胰岛素、HOMA-IR均升高,ISI均降低,且IH8组更明显(F=161.92、51.46、126.99、83.87,P均<0.05)。与对照组相比,IH4组及IH8组肝细胞GLUT-2、GCK蛋白表达均降低,且IH8组更显著(F=184.91、240.85,P均<0.05)。Pearson相关分析显示,GLUT-2、GCK平均灰度值与ISI呈负相关(r=-0.886、-0.906,P均<0.05),与HOMA-IR呈正相关(r=0.894、0.869,P均<0.05)。
间歇低氧暴露使大鼠肝细胞GLUT-2、GCK蛋白表达下调,可能参与间歇低氧条件下胰岛素抵抗的发生。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
阻塞性睡眠呼吸暂停低通气综合征(OSAHS)因对心、脑血管及代谢、内分泌等的影响已成为一个日益严重的医疗问题。OSAHS与2型糖尿病(T2DM)常合并存在。研究发现,OSAHS是T2DM发生、发展的独立危险因素,多达40%的OSAHS患者合并T2DM,且随OSAHS严重程度的增加,T2DM发病率升高,该类患者血糖更加不易控制[1,2]。慢性间歇低氧是OSAHS最具特征的病理生理改变,可通过氧化应激、系统性炎性反应、交感神经激活等机制影响胰岛素信号通路,导致胰岛素抵抗(IR)[3]。但具体分子机制仍不明。葡萄糖转运蛋白(GLUT)-2、葡萄糖激酶(GCK)均特异表达于肝细胞,是胰岛素代谢通路的终末信号分子,其表达异常影响胰岛素的降糖效应[4]。本实验通过模拟OSAHS的间歇低氧状态,建立慢性间歇低氧大鼠模型,以GLUT-2、GCK为研究目标,探讨间歇低氧引起IR的相关分子机制,为相关临床和实验研究提供理论依据。
便携式测氧仪AX-300购自A Teledyne Technologies Company英国公司;血糖仪AW06336402B、血糖试纸购自美国强生医疗器械公司;KB3127A大鼠胰岛素ELISA试剂盒购自上海凯博生化试剂有限公司;MK3型全自动中文酶标仪购自上海雨晨生物技术有限公司;GLUT-2、GCK免疫组化一抗购自北京博奥森生物技术有限公司;SABC二步法免疫组化试剂盒购自武汉博士德生物工程有限公司;其他仪器包括石蜡切片机、烘片机(LEICA),氮气、氧气减压表(上海仪表厂)及电磁阀门JB-16(荷兰)。
选取24只健康雄性6周龄Sprague-Dawley(SD)大鼠,体重160~180 g,购自山西医科大学动物实验中心动物,许可证号:SCXK(晋)2009-0004,动物批号:SCXK(晋) 2009-0001。按随机数字表法将实验动物分为对照组、间歇低氧4周组(IH4组)和间歇低氧8周组(IH8组),每组8只。间歇低氧组(IH4组和IH8组)与对照组除通气模式不同外,其余实验条件均控制一致,对照组与IH4组于第4周末结束间歇低氧暴露,IH8组于第8周末结束间歇低氧暴露。
实验室自行研制的间歇低氧舱是由5 mm厚透明有机玻璃密封而成,舱体两侧预留通气孔用于保持舱内气压恒定,舱体大小65 cm×45 cm×50 cm,由单片机编制程序控制单向电磁阀按预设通气模式向舱内输送气体。通气模式设定:模型组:先充氮气45 s,使舱内氧浓度由21%逐渐降至6%~8%,持续10 s后切换为氧气,持续45 s使舱内氧浓度逐渐恢复至21%,再持续10 s,进入下一组循环。对照组两次充入气体均为压缩空气,循环模式同间歇低氧组。舱内安装测氧仪实时氧浓度监测。舱体内水分及CO2由生石灰吸收。暴露方案:于每日上午9:00至下午5:00将造模组置于间歇低氧舱,对照组置于间歇空气舱。期间可自由进食,饮水。
3组大鼠暴露结束后,禁食12 h,剪掉大鼠尾尖,用血糖仪配套试纸条蘸取尾尖血用于空腹血糖测定。使用10%水合氯醛腹腔注射麻醉大鼠,称重,沿腹正中线逐层分离各组织,暴露腹主动脉,真空管采血,静置后高速离心,取上清液于超低温(-70℃)冰箱保存备用。分离的肝组织在中性甲醛溶液中浸泡48 h,石蜡包埋、切片,以备免疫组织化学染色。
取尾尖静脉血滴于血糖仪配套试纸条上,读取数值。采用125I胰岛素放射免疫分析药盒,按操作说明检测超低温保存血清的胰岛素水平。
计算胰岛素敏感性指数(ISI)及稳态模型评估-胰岛素抵抗指数(HOMA-IR)。ISI=1/(空腹血糖×空腹胰岛素),HOMA-IR=(空腹血糖×空腹胰岛素)/22.5。
肝组织石蜡标本切片、脱蜡、水化,高温、高压抗原修复,抗原封闭,加兔抗鼠GLUT-2、GCK一抗(一抗稀释浓度分别为1∶100和1∶150),4℃过夜,复温,加山羊抗兔二抗,DAB染色,复染、封片。显微镜观察实验结果,光镜下采图,利用Image-Pro Plus图像软件测定灰度值,对实验结果进行定量分析。
实验结果采用SPSS 13.0统计软件进行分析,所得正态分布的计量资料以 ±s表示,多组间均数比较采用单因素方差分析,组间两两比较采用LSD-t检验,两变量相关性分析采用Pearson直线相关分析,P<0.05为差异有统计学意义。
±s表示,多组间均数比较采用单因素方差分析,组间两两比较采用LSD-t检验,两变量相关性分析采用Pearson直线相关分析,P<0.05为差异有统计学意义。
暴露结束后3组大鼠体重之间差异无统计学意义(P>0.05)。与对照组相比,IH4组、IH8组空腹血糖、空腹胰岛素及HOMA-IR升高,ISI降低(P均<0.05),且以IH8组变化更为明显(P<0.05),见表1。
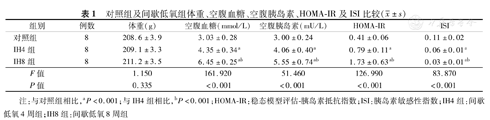
对照组及间歇低氧组体重、空腹血糖、空腹胰岛素、HOMA-IR及ISI比较( ±s)
±s)
对照组及间歇低氧组体重、空腹血糖、空腹胰岛素、HOMA-IR及ISI比较(±s)
| 组别 | 例数 | 体重(g) | 空腹血糖(mmol/L) | 空腹胰岛素(mU/L) | HOMA-IR | ISI | |
|---|---|---|---|---|---|---|---|
| 对照组 | 8 | 208.6±3.9 | 3.03±0.28 | 3.00±0.24 | 0.41±0.06 | 0.11±0.02 | |
| IH4组 | 8 | 209.1±3.3 | 4.35±0.34a | 4.06±0.40a | 0.79±0.11a | 0.06±0.01a | |
| IH8组 | 8 | 211.2±3.5 | 6.45±0.25ab | 5.55±0.74ab | 1.73±0.63ab | 0.03±0.01ab | |
| F值 | 1.150 | 161.920 | 51.460 | 126.990 | 83.870 | ||
| P值 | 0.335 | <0.001 | <0.001 | <0.001 | <0.001 | ||
注:与对照组相比,aP<0.001;与IH4组相比,bP<0.001;HOMA-IR:稳态模型评估-胰岛素抵抗指数;ISI:胰岛素敏感性指数;IH4组:间歇低氧4周组;IH8组:间歇低氧8周组
GLUT-2在大鼠肝细胞的细胞膜及细胞质中均有表达,主要分布于细胞膜,对照组可见细胞膜上密集的棕黄色颗粒分布;IH4组、IH8组细胞膜上棕黄色颗粒分布较对照组明显减少,与IH4组相比,IH8组变化更明显,且主要散在分布于细胞质,与IH4组相比,IH8组的棕黄色颗粒更少。GCK在肝细胞胞质、胞核均有表达,主要分布于细胞质,对照组可见肝细胞胞质广泛均匀分布的棕黄色颗粒,与对照组相比,IH4组、IH8组胞质中棕黄色颗粒减少,且不均匀。与IH4组相比,IH8组变化更显著。图像扫面定量分析显示,与对照组相比,IH4组及IH8组的GLUT-2、GCK平均灰度值逐渐升高,蛋白表达水平降低(P均<0.05)。与IH4组相比,IH8组GLUT-2、GCK表达更低(P均<0.05),见表2,图1(封3)。
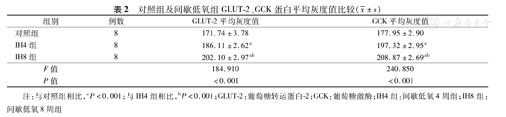
对照组及间歇低氧组GLUT-2、GCK蛋白平均灰度值比较( ±s)
±s)
对照组及间歇低氧组GLUT-2、GCK蛋白平均灰度值比较(±s)
| 组别 | 例数 | GLUT-2平均灰度值 | GCK平均灰度值 | |
|---|---|---|---|---|
| 对照组 | 8 | 171.74±3.78 | 177.95±2.90 | |
| IH4组 | 8 | 186.11±2.62a | 197.32±2.95a | |
| IH8组 | 8 | 202.10±2.97ab | 208.87±2.69ab | |
| F值 | 184.910 | 240.850 | ||
| P值 | <0.001 | <0.001 | ||
注:与对照组相比,aP<0.001;与IH4组相比,bP<0.001;GLUT-2:葡萄糖转运蛋白-2;GCK:葡萄糖激酶;IH4组:间歇低氧4周组;IH8组:间歇低氧8周组
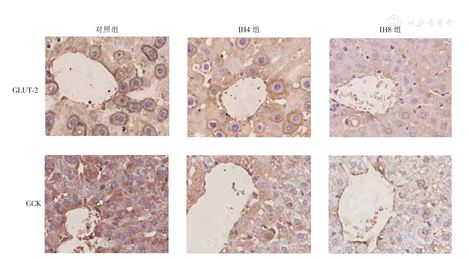
注:IH4组:间歇低氧4周组;IH8组:间歇低氧8周组;GLUT⁃2:葡萄糖转运蛋白⁃2;GCK:葡萄糖激酶
HOMA-IR与GLUT-2、GCK的平均灰度值呈正相关(r=0.894、0.869,P均<0.05),ISI与GLUT-2、GCK的平均灰度值呈负相关(r=-0.886、-0.906,P<均0.05)。
OSAHS患者由于睡眠过程中反复的咽部肌肉塌陷,上气道完全或不完全关闭,导致呼吸暂停和(或)低通气的发生。慢性间歇低氧是OSAHS最具特征的病理生理改变,而其诱发的氧化应激被认为是OSAHS引起多系统并发症的最重要机制[5]。尤其在IR及T2DM的发生、发展中起重要作用。OSAHS患者睡眠中反复发生呼吸暂停,使机体长期处于短暂高频的间歇低氧状态(氧饱和度持续下降15~60 s,继而上升,循环进行),类似缺血/再灌注损伤。缺氧/缺血期细胞适应低氧环境,复氧/再灌注导致细胞内氧突然增加,线粒体功能紊乱导致活性氧簇大量生成,肝细胞对缺氧有较好的适应能力,类似缺血/再灌注的肝损伤,缺氧期氧饱和度持续降低,细胞可适应低氧环境,而低氧后氧饱和度恢复过程即复氧期可致细胞内氧气量突然增加,频繁的低氧/复氧交替可造成线粒体功能紊乱,导致活性氧簇大量生成,且OSAHS患者同时存在抗氧化能力的降低,氧化与抗氧化失衡导致机体氧化损伤[6,7]。活性氧簇不可逆的氧化各种生物分子,并参与多条信号转导通路,通过改变胰岛素信号通路中多种功能蛋白的表达、结构及生物学功能,影响胰岛素信号转导以及胰岛素受体后信号通路,导致IR的发生。活性氧簇可诱导多种有害的促炎因子及炎性因子表达,参与炎性反应通路的激活,还可使低氧诱导因子-1表达增多,后者被认为与交感神经激活有关[8]。而系统性炎性反应、交感神经激活等也是导致IR发生的重要机制[3]。
IR是2型糖尿病的发病基础,肝脏IR主要表现为糖原合成能力降低[9]。肝脏是胰岛素敏感器官,也是胰岛素受体分布最密集的器官之一,受胰岛素严密调控,是葡萄糖稳态调节的一个重要器官。生理浓度的胰岛素在机体餐后血糖升高时主要通过激活肝脏、肌肉等组织胰岛素信号级联的葡萄糖转运及糖原合成过程的相关蛋白或酶,促进糖原合成而降低血糖[10]。GLUT-2、GCK负责外周葡萄糖摄取入肝及葡萄糖入胞后磷酸化过程,而后者代谢产物6-磷酸葡萄糖是葡萄糖代谢的中间产物,在机体血糖升高时主要进入糖原合成途径,促进合成糖原,两者表达异常,可使糖摄取及糖原合成过程受阻,导致IR的发生。
相关研究表明,肝脏胰岛素受体与GLUT-2在细胞膜上存在物理连接,这种耦合形成胰岛素调节肝葡萄糖转运的基础,具体相关机制仍不明[11]。因此,肝细胞膜上GLUT的表达量将直接影响葡萄糖转运过程。GLUT-2表达降低可造成葡萄糖转运入细胞过程受阻,葡萄糖摄取和利用率降低,导致IR。本实验显示,IH4组及IH8组GLUT-2表达较对照组减少,且随暴露时间延长效果更显著,GLUT-2的平均灰度值与HOMA-IR呈正相关,与ISI呈负相关,说明间歇低氧环境下GLUT-2表达减少,发生IR。
正常生理状态下,肝脏GCK活性受胰岛素调节,进餐后,胰岛素分泌增多,GCK活性增强,加速葡萄糖进入糖酵解及糖原合成,降低血糖[12]。GCK表达异常可使糖原合成水平及合成速率降低,导致肝脏IR。Seoane等[13]用Zuker糖尿病肥胖大鼠模型在研究T2DM时发现,模型组GCK表达较正常组下降40%,其糖原水平及合成速率均低于正常组。用含有GCK基因的腺病毒转染Zuker糖尿病肥胖大鼠使其过度表达GCK,结果其糖代谢恢复正常,糖原水平及合成速率与未转染模型鼠相比明显升高。本实验发现与对照组相比,IH4组及IH8组GCK表达明显减少,且IH8组更明显,GCK的平均灰度值与HOMA-IR呈正相关,与ISI呈负相关,说明慢性间歇低氧条件下GCK表达受阻,肝细胞处理葡萄糖的能力降低,呈现IR。
综上所述,慢性间歇低氧暴露可通过影响胰岛素代谢通路终末信号分子GLUT-2及GCK的蛋白表达,导致IR,引起机体糖代谢紊乱、胰岛素分泌异常,在T2DM发生、发展中起重要作用。而慢性间歇低氧诱导GLUT-2及GCK蛋白表达下降的同时是否存在其生物学功能异常,使慢性间歇低氧暴露大鼠过表达GLUT-2及GCK蛋白能否改善IR,仍需进一步探究。

























