先天性肾上腺皮质增生症(CAH)主要由类固醇生成酶基因突变或细胞色素P450氧化还原酶(POR)基因突变引起,是一种少见的遗传性代谢紊乱,与多囊卵巢综合征(PCOS)具有类似临床表现,常需进行鉴别诊断。本文报道了1例由POR基因突变导致的CAH患者,经基因检测证实其POR基因存在c.1370G>A(外显子12)纯合突变,诊断为细胞色素P450氧化还原酶缺陷症(PORD),提示临床上综合评估体格检查、肾上腺和性腺功能检查结果是识别PORD的关键,必要时需进行基因检测以明确诊断,继而进行精准治疗。


本刊2021年版权归中国全科医学杂志社所有
未经编辑部许可,不得任意转载和摘编
本刊所发表作品仅为作者观点,并不代表编委会和编辑部意见
如有印装质量问题请向本刊发行部调换
月经紊乱是育龄期女性至内分泌科门诊就诊的常见主诉,其病因复杂多样。多囊卵巢综合征(polycystic ovary syndrome,PCOS)是月经紊乱的常见病因之一,以雄激素升高及长期无排卵为主要特征,但由于PCOS与先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)具有类似临床表现而治疗方法及预后截然不同,因此临床医生在诊治以月经紊乱为主诉的患者时需对PCOS与CAH进行鉴别诊断。CAH是一种少见的遗传性代谢紊乱,也是一组常染色体隐性遗传病,主要由类固醇生成酶基因突变或细胞色素P450氧化还原酶(cytochrome P450 oxidoreductase,POR)基因突变引起[1]。本文报道了1例POR基因突变致CAH患者并进行了文献复习,以期提高临床对该病的认识与规范化诊治水平。
患者,女,16岁,足月顺产,因"月经紊乱4年余,末次月经后淋漓不尽1个月余"而于2020-01-11至安徽医科大学第一附属医院内分泌与代谢病科就诊。患者自诉2016年3月出现月经初潮,月经量较少,月经周期为3~4个月,经期为3~4 d,不伴痛经。2019-07-16,患者因"停经半年余"而至安徽医科大学第一附属医院门诊就诊,完善性激素六项检查发现睾酮为83 ng/dl(参考范围:14~76 ng/dl),余未见明显异常;行子宫及双附件超声检查提示双侧卵巢囊肿,考虑为高雄激素血症、PCOS(可能);予以炔雌醇环丙孕酮口服,治疗后患者月经规律来潮,2个月后自行停药,末次月经(2019-11-22)后开始淋漓不尽,遂再次就诊于安徽医科大学第一附属医院内分泌与代谢病科,门诊查17-羟孕酮为8.30 μg/L(参考范围:0.05~1.02 μg/L),以"高雄激素血症、PCOS(可能)"收入院。
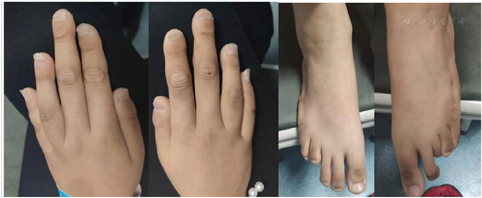
患者自诉发病以来意识清晰,精神可,无腹痛、乏力,二便正常,近2个月体质量增加约3 kg。患者为初中文化程度,学习成绩中上等;患者父亲48岁、身体健康,母亲44岁、身体健康,父母非近亲结婚,并均否认亲属有CAH相关疾病史;患者姐姐20岁,身体健康,14岁月经来潮,月经周期及月经量均正常。体格检查:血压122/74 mm Hg(1 mm Hg=0.133 kPa),身高165 cm、体质量46 kg、体质指数(BMI)16.9 kg/m2,指尖距166 cm、上部量82 cm、下部量83 cm,智力正常,全身皮肤较黑,头面部无畸形,甲状腺无肿大,腋毛等体毛发育正常,乳房Tanner分期Ⅱ期,阴毛呈女性分布、Tanner分期Ⅴ期,阴道及尿道分别开口,手指及脚趾均呈蜘蛛指样畸形并伴有指间关节肥大(图1),四肢及脊柱未见明显畸形及活动障碍,心、肺、腹部及神经系统查体均未见明显异常。
实验室检查:血常规、肝肾功能、血电解质及甲状腺功能检查均未见明显异常;性激素及肾上腺激素检查提示雄激素、17-羟孕酮、雄烯二酮水平升高,黄体生成素/促卵泡激素>2(表1);8:00促肾上腺皮质激素(ACTH)轻度升高(表2);进一步完善快速ACTH兴奋试验(分别于25 U ACTH静脉推注前及静脉推注后1 h抽取外周静脉血测定17-羟孕酮、皮质醇和孕酮)发现17-羟孕酮水平明显升高,但皮质醇水平升高不明显(表3)。子宫及双附件超声检查示子宫前位,大小为13 mm×31 mm×26 mm、形态规则,内膜居中、厚2 mm,左侧卵巢多个液暗区、最大为26 mm×47 mm,右侧卵巢有一23 mm×36 mm大小液暗区;子宫及双附件区未见明显异常血流信号。双侧肾上腺CT平扫+增强检查结果示左侧肾上腺内侧支稍增粗。
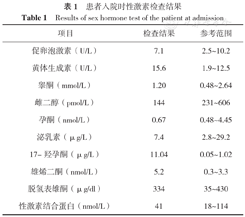
患者入院时性激素检查结果
Results of sex hormone test of the patient at admission
患者入院时性激素检查结果
Results of sex hormone test of the patient at admission
| 项目 | 检查结果 | 参考范围 |
|---|---|---|
| 促卵泡激素(U/L) | 7.1 | 2.5~10.2 |
| 黄体生成素(U/L) | 15.6 | 1.9~12.5 |
| 睾酮(mmol/L) | 1.20 | 0.48~2.64 |
| 雌二醇(pmol/L) | 144 | 231~606 |
| 孕酮(nmol/L) | 0.67 | 0.48~4.45 |
| 泌乳素(μg/L) | 7.4 | 2.8~29.2 |
| 17-羟孕酮(μg/L) | 11.04 | 0.05~1.02 |
| 雄烯二酮(nmol/L) | 5.2 | 0.3~3.3 |
| 脱氢表雄酮(μg/dl) | 334 | 35~430 |
| 性激素结合蛋白(nmol/L) | 41 | 18~114 |
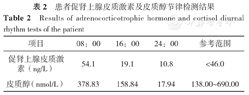
患者促肾上腺皮质激素及皮质醇节律检测结果
Results of adrenocorticotrophic hormone and cortisol diurnal rhythm tests of the patient
患者促肾上腺皮质激素及皮质醇节律检测结果
Results of adrenocorticotrophic hormone and cortisol diurnal rhythm tests of the patient
| 项目 | 08:00 | 16:00 | 24:00 | 参考范围 |
|---|---|---|---|---|
| 促肾上腺皮质激素(ng/L) | 54.1 | 19.1 | 10.8 | <46.0 |
| 皮质醇(nmol/L) | 378.83 | 158.84 | 17.94 | 138.00~690.00 |
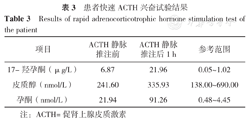
患者快速ACTH兴奋试验结果
Results of rapid adrenocorticotrophic hormone stimulation test of the patient
患者快速ACTH兴奋试验结果
Results of rapid adrenocorticotrophic hormone stimulation test of the patient
| 项目 | ACTH静脉推注前 | ACTH静脉推注后1 h | 参考范围 |
|---|---|---|---|
| 17-羟孕酮(μg/L) | 6.87 | 21.96 | 0.05~1.02 |
| 皮质醇(nmol/L) | 241.60 | 335.93 | 138.00~690.00 |
| 孕酮(nmol/L) | 21.94 | 91.26 | 0.48~4.45 |
注:ACTH=促肾上腺皮质激素
(1)患者因月经紊乱就诊,门诊结合患者病史、性激素六项和子宫及双附件超声检查结果拟诊"高雄激素血症、多囊卵巢综合征(可能)"而收入院;(2)患者皮肤较黑,入院后相关检查提示患者不仅存在高雄激素血症,而且肾上腺来源的雄激素前体明显增多并以17-羟孕酮水平升高为著,促肾上腺皮质激素(ACTH)偏高,因此不能排除先天性肾上腺皮质增生症(CAH);(3)患者无生殖器畸形,血电解质及血压均正常,仅表现为月经紊乱与卵巢囊肿,完善快速ACTH兴奋试验发现17-羟孕酮水平明显升高,同时实验室检查提示睾酮、雄烯二酮及ACTH升高,查体可见手指及脚趾均呈蜘蛛指样畸形,结合病史,考虑可能为非典型CAH;(4)取得患者及其家属知情同意后完善家系基因检测,检出细胞色素P450氧化还原酶(POR)基因c.1370G>A(外显子12)突变,确诊为细胞色素P450氧化还原酶缺陷症。
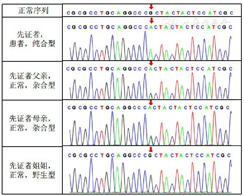
基因检测:充分与患者及其家属沟通病情并签署知情同意书后,留取患者及其父母、姐姐外周静脉血3 ml,采用高通量测序技术进行基因检测(北京智因东方),结果显示患者(先证者)POR基因存在c.1370G>A(外显子12)纯合突变〔在线《人类孟德尔遗传》(OMIM)号:613571〕,经Sanger测序验证,符合细胞色素P450氧化还原酶缺陷症(P450 oxidoreductase defeciency,PORD);患者父母均为POR基因杂合突变携带者,患者姐姐为POR基因野生型,均未见基因突变,符合常染色体隐性遗传规律(图2)。同时对患者CYP21A1基因进行检测,发现其IVS2-13位点为A的纯合形式,经在ClinVar数据库中验证为良性突变。
诊断及治疗:结合患者病史、临床表现及各项检查结果诊断为非典型CAH;完善家系基因检测,发现患者POR基因c.1370G>A(外显子12)突变,确诊为PORD;予以醋酸泼尼松片(5 mg/d)口服并准予出院。出院后患者月经正常,月经周期约为30 d;治疗期间因双侧臀部及大腿处皮肤出现紫纹、瘙痒而停药,待皮肤紫纹、瘙痒好转后继续用药,随访至今月经正常。
以"PORD""POR缺陷症""细胞色素P450氧化还原酶缺陷症"为关键词在中国知网及万方数据知识服务平台进行检索,共检索出相关文献13篇[2,3,4,5,6,7,8,9,10,11,12,13,14],涉及23例PORD确诊病例。23例PORD确诊病例男女比例为13/10,主要临床表现包括生殖器畸形、性激素异常、骨骼畸形、卵巢囊肿等,POR基因突变以R457H突变为主(20/46)。详见表4。
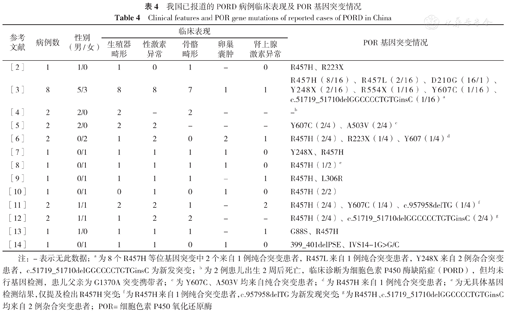
我国已报道的PORD病例临床表现及POR基因突变情况
Clinical features and POR gene mutations of reported cases of PORD in China
我国已报道的PORD病例临床表现及POR基因突变情况
Clinical features and POR gene mutations of reported cases of PORD in China
| 参考文献 | 病例数 | 性别(男/女) | 临床表现 | POR基因突变情况 | ||||
|---|---|---|---|---|---|---|---|---|
| 生殖器畸形 | 性激素异常 | 骨骼畸形 | 卵巢囊肿 | 肾上腺激素异常 | ||||
| [2] | 1 | 1/0 | 1 | 0 | 1 | - | 0 | R457H、R223X |
| [3] | 8 | 5/3 | 8 | 8 | 7 | 1 | 1 | R457H(8/16)、R457L(2/16)、D210G(16/1)、Y248X(2/16)、R554X(1/16)、Y607C(1/16)、c.51719_51710delGGCCCCTGTGinsC(1/16)a |
| [4] | 2 | 2/0 | 2 | - | 2 | - | - | -b |
| [5] | 2 | 2/0 | 2 | 2 | - | - | - | Y607C(2/4)、A503V(2/4)c |
| [6] | 2 | 0/2 | 1 | 2 | 0 | 2 | 1 | R457H(2/4)、R223X(1/4)、Y607(1/4)d |
| [7] | 1 | 0/1 | 1 | 1 | 1 | 1 | 0 | Y248X、R457H |
| [8] | 1 | 0/1 | 1 | 1 | 1 | 1 | 0 | R457H(1/2)e |
| [9] | 1 | 0/1 | 1 | 1 | 1 | - | 1 | R457H、L306R |
| [10] | 1 | 0/1 | 0 | 1 | 0 | 1 | 0 | R457H(2/2) |
| [11] | 2 | 1/1 | 2 | 2 | 1 | - | 2 | R457H(2/4)、Y607C(1/4)、c.957958delTG(1/4)f |
| [12] | 2 | 1/1 | 1 | 2 | 2 | - | - | R457H(2/4)、c.51719_51710delGGCCCCTGTGinsC(2/4)g |
| [13] | 1 | 1/0 | 1 | 1 | 1 | - | 1 | G88S、R457H |
| [14] | 1 | 0/1 | 1 | 1 | 0 | 1 | 0 | 399_401delPSE、IVS14-1G>G/C |
注:-表示无此数据;a为8个R457H等位基因突变中2个来自1例纯合突变患者,R457L来自1例纯合突变患者,Y248X来自2例杂合突变患者,c.51719_51710delGGCCCCTGTGinsC为新发突变;b为2例患儿出生2周后死亡,临床诊断为细胞色素P450酶缺陷症(PORD),但均未行基因检测,患儿父亲为G1370A突变携带者;c为Y607C、A503V均来自纯合突变患者;d为R457H来自1例纯合突变患者;e为无具体基因检测结果,仅提及检出R457H突变;f为R457H来自1例纯合突变患者,c.957958delTG为新发现突变;g为R457H、c.51719_51710delGGCCCCTGTGinsC均来自2例杂合突变患者;POR=细胞色素P450氧化还原酶
由于类固醇合成酶17-α羟化酶(CYP17A1)、21-羟化酶(CYP21A2)等细胞色素P450(CYP450)酶活性的发挥均需POR进行电子转移[15],因此PORD患者临床表现复杂、易与其他疾病混淆并导致误诊。由于对PORD不完全了解及基因测序技术限制,POR基因突变相关疾病一直未被正确认识。2004年,FLÜCK等[16]首次报道了POR基因突变病例,并认为其应与17羟化酶缺陷症、21羟化酶缺陷症等相鉴别。
POR基因位于7号染色体长臂(7q11 2),由15个外显子组成,跨越大小约为329 kb的区域。目前已报道的POR基因突变达上百种,涉及错义、移码和剪接位点突变等,但该基因突变位点分散、无明显热点。多数已被鉴定的POR基因错义突变位于POR基因的中央电子转移区附近,易破坏电子转移过程。研究表明,PORD发病率无明显性别差异,但在高加索人群中A287P是最常观察到的突变,而在日本人群中R457H是最常观察到的突变[17]。
CYP450酶包括Ⅰ型酶和Ⅱ型酶两种,其中Ⅰ型酶位于线粒体外膜,包含细胞色素P450侧链裂解酶(cytochrome P450Scc,CYP11A1)、11-β羟化酶(CYP11B1)、醛固酮合酶(CYP11B2)等;Ⅱ型酶位于光滑内质网,包括CYP17A1、细胞色素P450芳香化酶(cytochrome P450arom,CYP19A1)、CYP21A2等[18]。PORD患者可因POR基因突变导致CYP450酶活性下降甚至消失并引起多种临床表现:CYP21A2活性下降可导致17羟孕酮在胎儿体内累积并通过"后门途径"(胎儿在母体中时可通过5α还原酶1将17羟孕酮转化为17羟-别孕酮并进一步生成雄酮、雄烯二醇,最后转化为最具生物活性的雄激素双氢睾酮,而由于该途径不属于传统的雄激素生成途径,因此被称为"后门途径";胎儿出生后5α还原酶1转化为5α还原酶2,"后门途径"关闭[19])转化为有活性的二氢睾酮;CYP19A1活性下降可导致胎盘雄激素向雌激素转化过程受阻及大量睾酮产生,并由于残留的POR活性而导致外生殖器男性化[20],同时过量的雄激素还可使母体出现雄性化表现,但会在产后迅速好转。此外,在CYP19A1活性下降胎儿出生后,由于"后门途径"被关闭、只留下经典雄激素生成途径[21],因此其血清雄激素水平会恢复正常或处于偏低水平。与其他类型CAH相比,PORD患者可见两性外生殖器畸形,其中女孩出现明显的外生殖器男性化表明出生前雄激素过多,而男孩可能表现为男性化不足且程度不一(从交界性小阴茎到严重的会阴、阴囊下裂均可能发生)。
PORD患者成年后主要表现为性激素合成障碍,并以青春期发育延迟为主要特征。本例患者因月经紊乱而就诊,查体发现乳房发育迟缓。王春庆等[8]曾报道1例因巨大卵巢囊肿就诊并被确诊的PORD患者,本例患者存在双侧卵巢囊肿,但其卵巢囊肿原因主要分为以下两个方面:(1)雌激素缺乏导致促卵泡激素及黄体生成素分泌增加,继而刺激卵巢增生;(2)POR缺乏引起羊毛甾醇14α-去甲基化酶(CYP51A1)活性下降并导致促减数分裂甾醇减少,继而造成卵母细胞减数分裂及成熟障碍[22,23]。因此,临床发现疑似PORD病例时应注意与PCOS相鉴别。
在CAH的数种亚型中,仅PORD会出现骨骼畸形,且其表现类似于Antley-Bixler综合征(Antley-bixler syndrome,ABS)的表型,主要特征包括颅骨前突、面部中部发育不全及其他骨骼异常(包括手臂相邻骨骼融合、蜘蛛指样畸形)。目前,PORD导致骨骼畸形的具体机制尚不完全明确,可能与如下机制有关:PORD导致胆固醇生物合成受损尤其是依赖POR的CYP51A1活性受损、胆固醇减少、软骨细胞中Hedge-hog(Hh)表达降低[24],而Hh信号通路对胚胎发育(生长、形态发生)和骨形成过程的调节具有重要作用。PURSLEY等[25]研究表明,妊娠期接受氟康唑(一种已知的CYP51A1抑制剂)治疗的孕妇所娩出的婴幼儿表现为ABS样骨骼畸形。本例患者表现为蜘蛛指样畸形,但四肢活动自如、骨骼畸形较轻,提示不同类型的POR基因突变对相关酶活性的影响可能存在差异,而POR基因突变类型与临床表型间的关系尚需进一步研究。
需要指出的是,PORD患者可能会在新生儿筛查或基线生化检查中因血清CYP17A1水平升高而被发现,有时也会因CYP21A2基因突变而被误诊为21羟化酶缺陷症。KOYAMA等[26]基于9例PORD患者、21例21羟化酶缺陷症患者及67例短暂性CYP17A1升高者提出了一种两步生化检测方法以鉴别21羟化酶缺陷症与PORD,具体如下:留取受试者尿液标本,通过气相色谱/质谱分析尿液中类固醇排泄特点,之后先进行四氢可的松/三羟基孕酮(THEs/Ptl)比值测定以鉴别21羟化酶缺陷症与PORD、17羟化酶缺陷症与短暂性CYP17A1升高,再根据尿11β-羟基雄甾酮(11β-hydroxyandrosterone,11-HA)含量进一步鉴别21羟化酶缺陷症与PORD。KOYAMA等[26]提出的两步生化检测方法为难以开展基因检测者提供了新的诊断思路,但由于其研究仅在日本患者中开展,因此该方法对我国PORD患者的鉴别诊断价值尚需进一步研究证实。
综上所述,PORD属常染色体隐性遗传疾病,但POR基因的复杂突变谱导致PORD患者的临床表现多种多样,临床上若发现青春期女性月经紊乱或闭经、伴雄激素升高、伴或不伴特征性骨骼畸形,应考虑CAH的可能性并与PCOS、21羟化酶缺陷症进行鉴别、积极完善相关检查,而高度疑似PORD患者应进行基因检测以明确诊断,进而达到早期诊断、规范治疗、优生优育(对有生育计划者进行产前咨询及诊断)的目的。
本文无利益冲突。


























