
发育迟缓(DD)/精神发育迟缓(MR)病因复杂,临床表现多样、异质性强,该类患儿的早期精准诊断十分困难,目前国内鲜有大样本分析该类患儿的临床资料及基因检测结果。
分析DD/MR患儿基因检测结果,为DD/MR患儿确定遗传学诊断、制订治疗方案和判断预后提供依据。
选取2017年9月至2021年9月于昆明市儿童医院康复科就诊的致病原因尚不明确的93例DD/MR患儿为研究对象,对患儿进行全外显子组测序(WES)和拷贝数变异(CNV)检测,分析与患儿临床表现相关的致病性基因突变位点和CNV特点,分析基因突变检出情况。
93例患儿临床表现包括运动发育落后、智力低下或全面性发育落后,发育水平落后于正常发育里程碑。共检出遗传变异74例,检出率为79.6%,其中40例(43.0%)为致病性基因突变,13例(14.0%)为基因CNV,21例(22.6%)为突变意义未明。基因检测结果共涉及50种基因,所致疾病中SMN1基因突变引起的脊髓性肌萎缩症所占比例最高(10.0%,4/40),其次为COL6A2基因突变引起的Bethlem综合征1型(7.5%,3/40)及CSPP1基因突变所致的Joubert综合征21型(5.0%,2/40)。
致病基因突变和基因CNV可能是导致DD/MR的主要病因,SMN1、COL6A2、CSPP1为DD/MR患者常见突变基因,WES结合CNV检测对明确DD/MR的病因,特别是对诊断表型和临床表现不典型的患儿有重要意义。


本刊2023年版权归中国全科医学杂志社所有
未经编辑部许可,不得任意转载和摘编
本刊所发表作品仅为作者观点,并不代表编委会和编辑部意见
如有印装质量问题请向本刊发行部调换
发育迟缓(development delay,DD)/精神发育迟缓(mental retardation,MR)是儿童康复领域常见疾病。DD是指患儿存在明显的社会交往、运动、认知、语言等方面适应能力缺陷,运动、语言、认知等方面落后于正常儿童发育里程碑,可以是某一领域的发育落后,也可以累及多个领域引起全面性发育迟缓(global developmental delay,GDD)[1]。MR常见于18周岁以下的儿童,主要表现为智力低下及社会适应能力不足[2]。MR常用于诊断年龄≥5岁的智力低下的儿童,而DD/GDD用于诊断年龄<5岁,在运动、语言、认知单个或多个方面发育落后的儿童[3]。WHO报道DD/MR发病率为1%~3%[4],病因较为复杂,除遗传因素、环境因素外,内分泌异常、围生期因素等也会引起患儿发育落后,且该类患儿的临床表现多样、异质性强,这给DD/MR患儿的早期精准诊断带来了一定的难度[5]。全外显子组测序(whole exome sequencing,WES)和拷贝数变异(copy number variations,CNV)检测逐渐被用于对DD/MR患儿进行遗传学诊断。本文以2017年9月至2021年9月于昆明市儿童医院康复科就诊发病原因不明的DD/MR患儿为研究对象,分析了患儿的临床症状和基因突变情况,为发病原因不明的DD/MR的临床诊断提供理论依据。
选取2017年9月至2021年9月,于昆明市儿童医院康复科就诊的诊断为发病原因不明的DD/MR患儿作为研究对象。纳入病例至少符合以下两项标准中的一项:(1)存在运动、语言、认知等水平发育落后或全面性发育迟缓;(2)特殊面容。排除标准:(1)围生期感染、出生后中枢神经系统感染、缺氧、产伤、早产等其他已知原因造成的遗传综合征;(2)常见遗传代谢性疾病;(3)不同意进行基因检测;(4)失访。本研究通过昆明市儿童医院医学伦理委员会批准(批号:2017-03-269-K01),患儿监护人均已签署知情同意书。
收集患儿的病例资料,包括临床资料、发育评估量表评分、影像学检查、脑电图等辅助检查结果。
由本科室专业发育评估医师统一对纳入患儿进行神经发育评估。年龄<5岁的患儿采用儿童神经心理行为检查量表(简称儿心量表)[6]测定患儿的发育商(development quotient,DQ);年龄≥5岁的患儿采用中国韦氏智力量表测定患儿的智商(intelligence quotient,IQ)[7];采用婴儿-初中生社会适应性能力量表判定社会适应能力[8]。其中DQ/IQ值50~70为轻度发育落后,35~49为中度发育落后,<34为重度发育迟缓[9]。
抽取2~4 ml外周静脉血于抗凝管。使用血液基因组提取试剂盒(康为世纪)进行DNA提取,并使用荧光计和琼脂糖对样本做质量监测。采用IDT公司xGen® Exome Research Panel v1.0捕获探针,构建全外显子和基因组数据库,测序深度大于120×,覆盖度1×。在智因东方转化医学研究中心使用illumina公司测序平台进行第二代高通量测序。检测到的CNV变异按照2020年美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)发布的指南进行评级划分[10]。
最终纳入93例患儿,其中男54例(58.1%),女39例(41.9%)。最小年龄为8个月,最大年龄为11岁2个月,平均年龄(5.1±2.3)岁。诊断为DD的76例(81.7%),诊断为MR的17例(18.3%)。患儿起病年龄为6个月~1岁29例(31.2%),>1~3岁47例(50.5%),>3岁17例(18.3%)。患儿临床表现包括运动发育落后、智力低下或全面性发育落后。
经过对93例患儿进行发育评估,结果显示轻中度发育落后43例(46.2%),其中男25例(26.9%),女18例(19.4%);重度发育落后38例(40.9%),男29例(31.2%),女性9例(9.7%)。
93例患儿中74例存在遗传变异,检出率为79.6%,其中40例(43.0%)为致病性基因突变,13例(14.0%)为基因CNV,21例(22.6%)为突变意义未明。致病性基因突变40例中男27例(67.5%),女13例(32.5%);年龄6个月~1岁10例(25.0%),>1~3岁17例(42.5%),>3岁13例(32.5%)。
40例致病性基因突变患儿发育水平评估结果显示,均有不同程度的发育落后,其中轻度6例(15.0%)、中度23例(57.5%)、重度11例(27.5%)。40例患儿中伴有特殊面容、通贯掌等先天性畸形25例(62.5%),反复癫痫发作9例(22.5%),脑电图异常17例(42.5%),见表1。
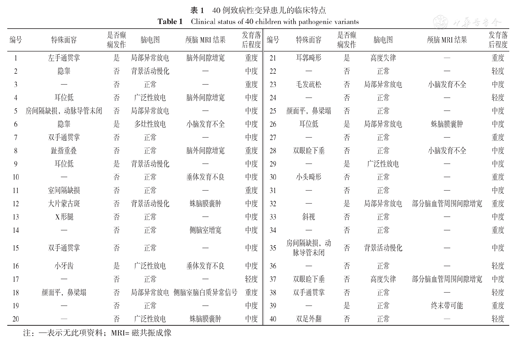
40例致病性变异患儿的临床特点
Clinical status of 40 children with pathogenic variants
40例致病性变异患儿的临床特点
Clinical status of 40 children with pathogenic variants
| 编号 | 特殊面容 | 是否癫痫发作 | 脑电图 | 颅脑MRI结果 | 发育落后程度 | 编号 | 特殊面容 | 是否癫痫发作 | 脑电图 | 颅脑MRI结果 | 发育落后程度 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 左手通贯掌 | 是 | 局部异常放电 | 脑外间隙增宽 | 重度 | 21 | 耳郭畸形 | 是 | 高度失律 | — | 重度 |
| 2 | 隐睾 | 否 | 背景活动慢化 | — | 中度 | 22 | — | 否 | 正常 | — | 轻度 |
| 3 | — | 否 | 正常 | — | 重度 | 23 | 毛发疏松 | 否 | 局部异常放电 | 小脑发育不全 | 中度 |
| 4 | 耳位低 | 否 | 广泛性放电 | 脑外间隙增宽 | 中度 | 24 | — | 否 | 正常 | — | 轻度 |
| 5 | 房间隔缺损,动脉导管未闭 | 否 | 局部异常放电 | — | 中度 | 25 | 颜面平,鼻梁塌 | 否 | 正常 | — | 中度 |
| 6 | 隐睾 | 是 | 多灶性放电 | 小脑发育不全 | 中度 | 26 | 耳位低 | 是 | 局部异常放电 | 蛛脑膜囊肿 | 中度 |
| 7 | 双手通贯掌 | 否 | 正常 | — | 中度 | 27 | — | 否 | 正常 | — | 重度 |
| 8 | 趾指重叠 | 否 | 正常 | 脑外间隙增宽 | 重度 | 28 | 双眼睑下垂 | 否 | 正常 | 小脑发育不全 | 中度 |
| 9 | 耳位低 | 是 | 背景活动慢化 | — | 中度 | 29 | — | 是 | 广泛性放电 | — | 中度 |
| 10 | — | 否 | 正常 | 垂体发育不良 | 中度 | 30 | 小头畸形 | 否 | 正常 | — | 重度 |
| 11 | 室间隔缺损 | 否 | 正常 | — | 重度 | 31 | — | 否 | 正常 | — | 中度 |
| 12 | 大片蒙古斑 | 否 | 背景活动慢化 | 蛛脑膜囊肿 | 中度 | 32 | — | 是 | 局部异常放电 | 部分脑血管周围间隙增宽 | 重度 |
| 13 | X形腿 | 否 | 正常 | — | 中度 | 33 | 斜视 | 否 | 正常 | — | 中度 |
| 14 | — | 否 | 正常 | 侧脑室增宽 | 中度 | 34 | — | 否 | 正常 | — | 重度 |
| 15 | 双手通贯掌 | 否 | 正常 | — | 中度 | 35 | 房间隔缺损,动脉导管未闭 | 否 | 背景活动慢化 | — | 中度 |
| 16 | 小牙齿 | 是 | 广泛性放电 | 垂体发育不良 | 中度 | 36 | — | 否 | 正常 | — | 轻度 |
| 17 | — | 否 | 正常 | — | 轻度 | 37 | 双眼睑下垂 | 否 | 高度失律 | 部分脑血管周围间隙增宽 | 中度 |
| 18 | 颜面平,鼻梁塌 | 否 | 局部异常放电 | 侧脑室脑白质异常信号 | 重度 | 38 | 双手通贯掌 | 否 | 正常 | — | 轻度 |
| 19 | — | 否 | 正常 | — | 中度 | 39 | — | 是 | 正常 | 终末带可能 | 重度 |
| 20 | — | 否 | 广泛性放电 | 蛛脑膜囊肿 | 中度 | 40 | 双足外翻 | 否 | 正常 | — | 轻度 |
注:—表示无此项资料;MRI=磁共振成像
93例患儿基因诊断结果包括以下基因:SMN1(3例)、COL6A2(3例)、CSPP1(2例),以下基因均为1例:MECP2、FOXG1、G6PD、LAMA2、GNB1、SHANK3、CHKB、KIAA0195、SPEG、RPGRIP1、CABP4、MCM3AP、SPR、GNAO1、SMARCA4、SYT1、CTCF、PAX3、CSNK2A1、TRIP12、PUF60、EP300、CNOT3、TCF4、NR2F1、NFIX、COL1A1、COL12A1、SPTAN1、FLNB、CHD8、AIFM1、SOS1、DVL1、NLGN3、RYR1、chrX、ALG8、AFF2、NLGN4X、QARS1、UBE3A、AGRN、CACNA1H、CABP4、OCA2、CACNA1C。其中SMN1基因突变引起的脊髓性肌萎缩症所占比例最高,共4例(10.0%),其次为COL6A2基因突变引起的Bethlem综合征1型3例(7.5%)及CSPP1基因突变所致的Joubert综合征21型2例(5.0%),见表2。13例(15.5%)基因CNV的基因变异位点见表3。

40例检出致病基因突变的遗传学分析
Genetic analysis of pathogenic gene mutations detected in 40 cases
40例检出致病基因突变的遗传学分析
Genetic analysis of pathogenic gene mutations detected in 40 cases
| 编号 | 基因 | 遗传方式 | 突变来源 | ACMG等级 | 突变方式 | 相关疾病 |
|---|---|---|---|---|---|---|
| 1 | SMN1 | AR | 父亲+母亲(杂合) | 1类-致病突变 | chr5/7、8号外显子纯合缺失 | 脊髓性肌萎缩症 |
| 2 | SMN1 | AR | 父亲(x1)—母亲(x1) | 1类-致病突变 | chr5/7、8号外显子纯合缺失 | 脊髓性肌萎缩症 |
| 3 | SMN1 | AR | 母亲(杂合) | 1类-致病突变 | chr5/7、8号外显子纯合缺失 | 脊髓性肌萎缩症 |
| 4 | SMN1 | AR | 父亲+母亲(杂合) | 1类-致病突变 | chr5/7、8号外显子纯合缺失 | 脊髓性肌萎缩症 |
| 5 | COL6A2 | AR/AD | 父亲(杂合) | 1类-致病突变 | chr21:47552017/c.2611G>A(p.D871N) | Bethlem综合征1型 |
| 6 | COL6A2 | AR/AD | 母亲(杂合) | 2类-可能致病 | chr21:47545383/c.1821T>A(p.C607*) | Bethlem综合征1型 |
| 7 | COL6A2 | AR/AD | 父亲(杂合) | 1类-致病突变 | chr21:47552017/c.2611G>A(p.D871N) | Bethlem综合征1型/先天性肌硬化/Ullrich先天性肌营养不良1型 |
| 8 | CSPP1 | AR | 父亲+母亲(杂合) | 2类-可能致病 | chr8:68074049/c.2527_2528delAT(p.M843Efs*25) | Joubert综合征21型 |
| 9 | CSPP1 | AR | 父亲+母亲(杂合) | 2类-可能致病 | chr8:68074049/c.2527_2528delAT(p.M843Efs*25) | Joubert综合征21型 |
| 10 | MECP2 | XL | 新发 | 1类-致病突变 | chrX:153296363/c.952C>T(p.R318C) | Rett综合征 |
| 11 | FOXG1 | AD | 新发 | 1类-致病突变 | chr14:29236986/c.506del(p.G169Afs*23) | 先天性变异型Rett综合征 |
| 12 | G6PD | XL | 母亲(杂合) | 1类-致病突变 | c.487G>A(p.G163S) | G6PD缺乏症 |
| 13 | LAMA2 | AR | 父亲(杂合) | 1类-致病突变 | chr6:129785589/c.7147C>T(p.R2383*) | Merosin缺乏或部分缺乏型先天性肌营养不良/肢带型肌营养不良23型 |
| 14 | GNB1 | AD | 新发 | 2类-可能致病 | chr1:1737957/c.224A>G(p.Q75R) | 精神发育迟缓42型 |
| 15 | SHANK3 | AD | 新发 | 2类-可能致病 | chr22:51159010/c.2797del(p.R933Dfs*39) | Phelan-McDermid综合征 |
| 16 | CHKB | AR | 母亲(杂合) | 2类-可能致病 | c.865dupA(p.T289Nfs*36) | 先天性肌营养不良Megaconial型 |
| 17 | KIAA0195 | AR | 父亲(杂合) | 2类-可能致病 | chr17:73494529/c.3643G>A(p.D1215N) | 智力发育障碍伴心脏缺陷和面部畸形 |
| 18 | SPR | AR/AD | 父亲+母亲(杂合) | 2类-可能致病 | chr2:73118535/c.655C>T(p.R219*) | 墨蝶呤还原酶缺乏型多巴反应性肌张力障碍 |
| 19 | COL1A1 | AD | 母亲(杂合) | 2类-可能致病 | c.3134dupC(p.G1046Wfs*20) | 成骨不全合并Ehlers-Danlos综合征1型 |
| 20 | COL12A1 | AD | 父亲(杂合) | 1类-致病突变 | c.4957+1G>A | Bethlem综合征2型 |
| 21 | MCM3AP | AR | 母亲(杂合) | 2类-可能致病 | c.5262A>C(p.R1754S) | 周围神经病变可伴智力发育受损 |
| 22 | GNAO1 | AD | 新发 | 1类-致病突变 | chr16:56370675/c.626G>A(p.R209H) | 发育性和癫痫性脑病17型/神经发育异常伴不自主运动 |
| 23 | SMARCA4 | AD | 新发 | 2类-可能致病 | chr19:11132545/c.2761C>T(p.L921F) | Coffin-Siris综合征4型 |
| 24 | SYT1 | AD | 新发 | 1类-致病突变 | chr12:79842738/c.1103T>C(p.I368T) | 贝克-戈登综合征 |
| 25 | CTCF | AD | 新发 | 2类-可能致病 | chr16:67645347/c.615_618delGAAA(p.K206Pfs*15) | 精神发育迟缓21型 |
| 26 | PAX3 | AD/AR | 新发 | 1类-致病突变 | chr2:223086101/c.798G>A(p.W266*) | 颅面-耳聋-手综合征/Waardenburg综合征1型/Waardenburg综合征3型 |
| 27 | CSNK2A1 | AD | 新发 | 2类-可能致病 | chr20:472969/c.548_550del(p.H183del) | Okur-Chung神经发育综合征 |
| 28 | TRIP12 | AD | 新发 | 1类-致病突变 | chr2:230650527/c.4814_4815del(p.V1605Dfs*16) | 智力障碍49型 |
| 29 | PUF60 | AD | 新发 | 2类-可能致病 | chr8:144899885/c.882_885del(p.Q295Tfs*63) | Verheij综合征 |
| 30 | EP300 | AD | 新发 | 2类-可能致病 | chr22:41573960/c.6245del(p.A2082Vfs*52) | Rubinstein-Taybi综合征2型—Menke-Hennekam综合征2型 |
| 31 | CNOT3 | AD | 新发 | 2类-可能致病 | chr19:54647786/c.303A>T(p.K101N) | 智力发育障碍伴语言迟缓 |
| 32 | TCF4 | AD | 新发 | 1类-致病突变 | chr18:52942977/c.662del(p.H221Lfs*13) | Fuchs角膜内皮营养不良3型—Pitt-Hopkins综合征 |
| 33 | NR2F1 | AD | 新发 | 2类-可能致病 | chr5:92921093/c.364T>C(p.C122R) | Bosch-Boonstra-Schaaf视神经萎缩综合征 |
| 34 | NFIX | AD | 新发 | 1类-致病突变 | chr19:13189458/c.988delG(p.D330Tfs*71) | Marshall-Smith综合征/Sotos综合征2型 |
| 35 | AFF2 | XL | 新发 | 2类-可能致病 | c.926T>C(p.I309T) | 精神发育迟缓FRAXE型 |
| 36 | NLGN4X | XL | 母亲(杂合) | 1类-致病突变 | c.1591G>A(p.A531T) | Asperger综合征易感性2型 |
| 37 | SPEG | AR | 母亲(杂合) | 2类-可能致病 | chr2:220333344/c.3187G>A(p.A1063T) | 中央核肌病 |
| 38 | RPGRIP1 | AR | 父亲(杂合) | 2类-可能致病 | chr14:21793098/c.2084A>G(p.Q695R) | 视锥视杆细胞营养不良13型/Leber先天性黑矇6型 |
| 39 | CABP4 | AR | 母亲(杂合) | 2类-可能致病 | chr11:67223662/c.370C>T(p.R124C) | 先天性非进行性视锥视杆细胞突触异常 |
| 40 | UGT1A1 | AR | 父亲(杂合) | 2类-可能致病 | chr2:234669144/c.211G>A(p.G71R) | Crigler-Najjar综合征Ⅰ型 |
注:ACMG=美国医学遗传学与基因组学学会,AR=常染色体隐性遗传,AD=常染色体显性遗传,XL=X染色体连锁遗传,G6PD=葡萄糖-6-磷酸脱氢酶;x1表示拷贝数为1
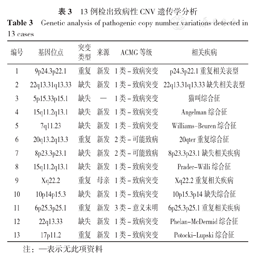
13例检出致病性CNV遗传学分析
Genetic analysis of pathogenic copy number variations detected in 13 cases
13例检出致病性CNV遗传学分析
Genetic analysis of pathogenic copy number variations detected in 13 cases
| 编号 | 基因位点 | 突变类型 | 来源 | ACMG等级 | 相关疾病 |
|---|---|---|---|---|---|
| 1 | 9p24.3p22.1 | 重复 | 新发 | 1类-致病突变 | p24.3p22.1重复相关表型 |
| 2 | 22q13.31q13.33 | 缺失 | 新发 | 1类-致病突变 | 22q13.31q13.33缺失相关表型 |
| 3 | 5p15.33p15.1 | 缺失 | — | 1类-致病突变 | 猫叫综合征 |
| 4 | 15q11.2q13.1 | 缺失 | 新发 | 1类-致病突变 | Angelman综合征 |
| 5 | 7q11.23 | 缺失 | 新发 | 1类-致病突变 | Williams-Beuren综合征 |
| 6 | 20q13.2q13.3 | 重复 | 新发 | 2类-可能致病 | 20qter重复综合征 |
| 7 | 8p23.3p23.1 | 缺失 | 新发 | 2类-可能致病 | 8p23.3p23.1缺失相关疾病 |
| 8 | 15q11.2q13.1 | 缺失 | 新发 | 1类-致病突变 | Prader-Willi综合征 |
| 9 | Xq22.2 | 重复 | 母亲 | 1类-致病突变 | Xq22.2重复相关疾病 |
| 10 | 10p14p15.3 | 缺失 | 新发 | 1类-致病突变 | 10p15.3p14缺失综合征 |
| 11 | 6p25.3p25.1 | 重复 | 新发 | 3类-意义未明 | 6p25.3p25.1重复相关疾病 |
| 12 | 22q13.33 | 缺失 | 新发 | 1类-致病突变 | Phelan-McDermid综合征 |
| 13 | 17p11.2 | 重复 | 新发 | 1类-致病突变 | Potocki-Lupski综合征 |
注:—表示无此项资料
DD/MR是常见的发育障碍性疾病,这类患儿的认知功能和社会适应能力明显落后于同龄正常儿童[11],文献报道DD/MR的病因中有遗传因素的占17.4%~47.1%[12],本研究43.1%的诊断阳性率与其一致。对不明原因导致的DD/MR患儿诊断比较困难,尤其是无特殊临床表型、特殊面容、先天性残疾的患儿,容易造成漏诊或误诊。早期干预对于DD/MR的患儿具有重要意义,可以最大限度地降低发育落后导致的功能障碍和生长发育方面的负面影响,可能帮助患儿重返家庭和社会[13]。然而仅少数患儿能够做到早期发现、早期识别及早期干预,发育落后患儿大多数在出生时无特殊症状,在发育过程中才逐渐出现发育落后的问题,导致错过最佳干预时机。因此,早期发现和诊断对DD/MR患儿十分重要。
DD/MR的症状复杂,表型异质性较大,很难通过一种特征性临床表现来确定是否与基因突变或染色体异常相关。此外影响患儿临床表现的遗传因素也有多种,包括突变基因位点的功能、缺失片段的大小、缺失片段所含的基因等。目前推测大量与DD/MR相关的基因突变和CNV尚未被发现,已发现的变异也需深入研究[14]。因此仍需通过大样本的遗传学研究进一步筛选出可能导致DD/MR的遗传学标志物。目前,已有研究将基因检测应用于无明确发病原因DD/MR的遗传学病因诊断中,美国相关指南指出,基因检测是诊断DD/MR的标准化流程之一[15]。基因检测中WES可以检测单个基因位点的改变,为诊断单基因导致的DD/MR提供依据,CNV可检测拷贝片段的复制、缺失、倒置等变异,分析变异与患者临床表型之间的关系。
2017年9月至2021年9月,于本院康复科就诊的DD/MR患儿共296例,主要通过出生史、生长发育史及辅助检查进行分析,其中有128例患儿有与其临床表现相关的病因,主要原因有早产导致低出生体质量、新生儿缺氧窒息、新生儿缺氧、缺血性脑病(HIE)、高胆红素血症、颅内感染或出血、脑回畸形、孤独症谱系障碍、甲状腺功能减退症、脑创伤等。本研究对无明确发病原因且同意进行基因检测的93例不明原因DD/MR患儿进行基因检测,发现脊髓性肌萎缩症、Bethlem综合征1型、Joubert综合征21型检出率较高。脊髓性肌萎缩症是一种常染色体隐性遗传神经元变性疾病,由运动神经元存活基因缺陷导致患者脊髓前角神经元细胞功能失调,最终引起进行性肌肉萎缩、无力[16]。Bethlem综合征是一种常染色体显性遗传性肌病,由COL6A1基因变异所致,主要为近端肌无力、长手指屈肌、手肘及脚踝等关节挛缩等[17]。Joubert综合征是一种神经系统罕见病,常表现为新生儿期阵发性呼吸暂停、小脑蚓部发育不良、肌力弱、发育落后等症状,文献报道了34个与之相关的致病基因,CSPP1是第21个被发现的致病基因[18]。40例致病性变异患儿中,伴有特殊面容、先天性畸形25例,主要表现为双手通贯掌、隐睾、耳位低、小牙齿、颜面平、鼻梁塌、斜视、心脏疾病、趾指重叠、X形腿、耳郭畸形、毛发疏松、共同性外斜视、眼球震颤等,反复癫痫发作9例,脑电图异常17例;患儿的发育水平评估结果均显示为不同程度的发育落后,同时发现不同的患儿可能存在同一基因片段缺失或重复、同一基因突变,但由于临床异质性,患儿的临床表型也存在一定差异。因CNV的基因变异位点关联的疾病包括猫叫综合征、Angelman综合征、Williams-Beuren综合征、Prader-Willi综合征、Phelan-McDermid综合征、Potocki-Lupski综合征。
综上所述,对DD/MR进行准确的病因学诊断,是临床进行有效康复治疗的前提,能最大限度减轻患儿功能障碍程度、改善患儿预后。基因突变和基因CNV是导致DD/MR发生的主要遗传学病因,采用WES结合CNV的方法能对明确DD/MR的病因提供依据,依据检测结果进行遗传咨询和针对性的治疗,提高疗效改善预后。在今后的研究中,可进一步进行采用WES结合CNV的方法发现DD/MR新的候选基因,为确定遗传学诊断、制订治疗方案和判断患儿预后提供依据。
本文无利益冲突。
王静,刘芸,黄浩宇,等.不明原因发育迟缓/精神发育迟缓儿童基因检测结果研究[J].中国全科医学,2023,26(8):933-938. [www.chinagp.net]
WANG J,LIU Y,HUANG H Y,et al. Developmental delay/mental retardation of unknown origin in children: genetic analysis of 93 cases[J]. Chinese General Practice,2023,26(8):933-938.

























