
毛发鼻指(趾)骨综合征(TRPS)是一种罕见的常染色体显性遗传病,其临床特征主要为毛发稀疏、梨状鼻、部分指(趾)骨锥形骨骺及短指畸形等。本文报道了1例因矮小、特殊面容于2021-01-07在石河子大学医学院第一附属医院儿科确诊为TRPS合并生长激素缺乏症的患儿,回顾性分析了患儿的临床资料及诊断过程,基因检测显示其TRPS1_ex6 c.2725dupT(p.Cys909Leufs*42)杂合移码突变为新发变异,根据美国医学遗传学与基因组学会(ACMG)指南判读为致病变异,确诊为TRPS合并生长激素缺乏症,并给予重组人生长激素治疗,随访1年2个月发现,重组人生长激素治疗可以改善TRPS合并生长激素缺乏症患儿的身高,且未引起患儿颅内压增高、甲状腺功能减退等不良反应。


本刊2023年版权归中国全科医学杂志社所有
未经编辑部许可,不得任意转载和摘编
本刊所发表作品仅为作者观点,并不代表编委会和编辑部意见
如有印装质量问题请向本刊发行部调换
毛发鼻指(趾)骨综合征(tricho-rhino-phalangeal syndrome,TRPS)是一种罕见的常染色体显性遗传病,1966年由瑞士儿科放射科医师GIEDION[1]首次描述,其主要特征为头发稀疏、生长缓慢(额颞叶区域明显),侧面眉毛稀疏,梨状鼻,人中长而平,上唇薄且边缘呈朱红色,耳朵突出及部分中节指(趾)关节弯曲、粗大等[2,3];最典型的放射学表现为锥形骨骺(CSE)[2],主要位于中指骨(通常情况下在2岁之前是检测不到的,但在某些情况下,在生命的第一年可以检测到轻度干骺端凸起),骨龄总是落后于实际年龄,直到青春期加速。TRPS合并生长激素缺乏症(growth hormone deficiency,GHD)使用重组人生长激素(recombinant human growth hormone,rhGH)治疗在国外罕见报道[4,5],疗效评价不一。本文回顾性分析1例确诊为TRPS合并GHD患儿的临床资料及诊断过程,并观察rhGH治疗TRPS合并GHD患儿的疗效及安全性,旨在提高临床医师对TRPS的认识。本研究已通过石河子大学医学院第一附属医院伦理委员会审批(KJ2021-134-01)。
患儿,女,3岁9个月,因发现生长迟缓2年于2021-01-07就诊于石河子大学医学院第一附属医院儿科。患儿系第二胎、第二产,足月剖宫产,出生时无窒息史。出生体质量3.2 kg,身长50 cm,新生儿期健康状况可,出生后母乳喂养,无体弱多病,2个月抬头,4个月翻身,6个月独坐,10个月会说话,13个月会走,智能发育无落后。母亲身高170 cm,父亲身高185 cm。父母、祖父母及外祖父母外貌、智力均正常。父母非近亲婚配,无家族遗传病史,患儿有一姐姐,目前10岁,身高145 cm,青春期女孩,发育及智力正常。
体格检查:身高95 cm(P3),体质量16.0 kg(P50),头围49.5 cm,指尖距94.1 cm,上部量51.8 cm,下部量45.2 cm,BMI 17.73 kg/m2;意识清晰,精神反应可,能正确对答;背部可见一牛奶咖啡斑,其余部位未见皮疹及出血点,浅表淋巴结无肿大,头发颜色偏黄、稀疏分布,前额饱满,眉毛色淡,睫毛细短,梨状鼻,无颈蹼,咽无充血,胸廓呈鸡胸,双侧肋外翻;心率约105次/min,律齐,心音有力,未闻及病理性杂音,呼吸规律,双肺呼吸音清,未闻及干湿性啰音;腹软,肝脾未触及肿大,无压痛及反跳痛,肠鸣音可。四肢脊柱无畸形,双侧手指短粗,双侧4、5中节指间关节略弯曲,不伴疼痛,无关节过伸;双乳B1期,外阴呈幼女型,未见阴毛及腋毛生长。
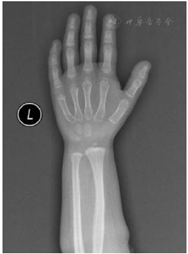
实验室及影像学检查:血常规、肝肾功能、电解质、心肌酶、血脂、血糖未见异常;甲状腺功能3项〔促甲状腺激素(TSH)、游离甲状腺素(FT4)、游离三碘甲状腺原氨酸(FT3)〕未见异常;促肾上腺皮质激素、皮质醇、17羟孕酮未见异常;染色体核型分析:46XX;微量元素6项(锌、铁、钙、镁、铜、铅)未见异常;25-羟基维生素D 89.63 μg/L。精氨酸联合左旋多巴激发试验生长激素(GH)峰值3.87 μg/L(>10 μg/L为正常),胰岛素样生长因子1(IGF-1)104 mg/L,胰岛素样生长因子结合蛋白3(IGFBP-3)3.62 mg/L。心脏彩超提示:房间隔卵圆孔未闭0.34 cm。肝胆胰脾双肾未见异常,肾上腺B超未见异常。左手腕骨X线片示:骨龄2岁,CSE伴指骨弥漫性缩短(图1)。脊柱正侧位片示:脊柱生理曲度可,无明显侧弯。颅脑+垂体磁共振成像(MRI)无异常。
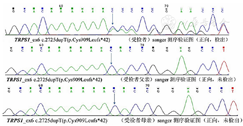
基因检测:根据患儿身材矮小、特殊面容,除GHD外,需考虑遗传代谢疾病的可能,取得患儿父母的知情同意后,抽取患儿及其父母外周血各2 mL,乙二胺四乙酸(EDTA)抗凝,送杭州迪安医学检验中心行全外显子测序及Sanger测序验证。结合患儿临床资料,对患儿的AARS,ANO5,CAPN3等共20 000个遗传性疾病基因组合外显子及其邻近±10 bp内含子进行了高通量检测和分析,并着重关注和分析了遗传代谢疾病、矮小症、生长发育迟缓、内分泌代谢系统相关的基因。结果显示,本次检测检出1个致病变异,TRPS1_ex6 c.2725dupT(p.Cys909Leufs*42),杂合移码突变;该移码突变会导致第909位氨基酸由半胱氨酸替换为亮氨酸并改变阅读框,在下游第41位密码子处产生终止密码子,使编码蛋白序列提前终止,产生截短蛋白或被降解,可能会对蛋白质的结构和功能产生较大影响;ClinVar数据库收录该变异位点为致病变异[6];该位点在正常东亚人群中的频率未见报道;经Sanger测序验证,患儿父母均未检出该变异,即患儿的该变异为新发变异;根据美国医学遗传学与基因组学会(ACMG)指南[7]判读为致病性变异,确诊为TRPS。患儿TRPS1_ex6 c.2725dupT(p.Cys909Leufs*42)发生杂合移码突变;其父母该位点均为正常基因。患儿及父母TRPS1基因突变序列见图2。
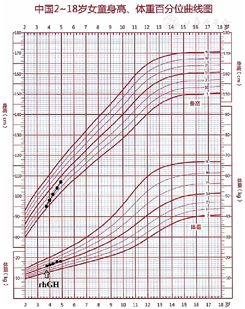
诊断及治疗:结合患儿身高处于同年龄、同性别P3百分位,生长速率<5 cm/年,骨龄落后实际年龄1.5岁,精氨酸联合左旋多巴激发试验GH峰值3.87 μg/L<5 μg/L,根据2008年矮身材儿童诊治指南[8],此患儿确诊为GHD。经家长同意给予患儿rhGH 0.100 U·kg-1·d-1皮下注射治疗,开始治疗时年龄为3岁9个月,身高95 cm,身高标准差积分(HtSDS)为-2 SD;治疗同时配合饮食、运动、睡眠指导及干预,每3个月定期复诊一次,监测血常规、肝肾功能、空腹血糖、胰岛素、C肽、甲状腺功能3项、IGF-1、IGFBP-3,监测生长速率及有无不良反应发生。结果显示,rhGH治疗1年2个月后,身高为107 cm,HtSDS为-1 SD,身高增长12 cm,年身高增长10.3 cm;随着治疗时间延长,IGF-1、IGFBP-3水平也逐渐升高;治疗过程中未出现颅内压增高、糖代谢异常、甲状腺功能减退等不良反应,见表1、图3。
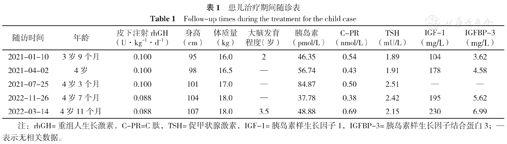
患儿治疗期间随诊表
Follow-up times during the treatment for the child case
患儿治疗期间随诊表
Follow-up times during the treatment for the child case
| 随访时间 | 年龄 | 皮下注射rhGH (U·kg-1·d-1) | 身高(cm) | 体质量(kg) | 大脑发育程度(岁) | 胰岛素(pmol/L) | C-PR (nmol/L) | TSH (mU/L) | IGF-1 (mg/L) | IGFBP-3 (mg/L) |
|---|---|---|---|---|---|---|---|---|---|---|
| 2021-01-10 | 3岁9个月 | 0.100 | 95 | 16.0 | 2 | 46.35 | 0.54 | 1.89 | 104 | 3.62 |
| 2021-04-02 | 4岁 | 0.100 | 98 | 16.5 | — | 56.74 | 0.43 | 1.91 | 178 | 4.58 |
| 2021-07-25 | 4岁3个月 | 0.100 | 101 | 17.0 | — | 84.87 | 0.50 | 2.51 | — | — |
| 2022-11-26 | 4岁7个月 | 0.088 | 104 | 18.0 | — | 37.78 | 0.38 | 2.42 | 195 | 5.62 |
| 2022-03-14 | 4岁11个月 | 0.088 | 107 | 18.0 | 3.5 | 48.88 | 0.69 | 2.15 | 230 | 6.99 |
注:rhGH=重组人生长激素,C-PR=C肽,TSH=促甲状腺激素,IGF-1=胰岛素样生长因子1,IGFBP-3=胰岛素样生长因子结合蛋白3;—表示无相关数据。
TRPS1是一种体内外序列特异性转录抑制因子,介导多种组织的细胞分化,TRPS1基因定位于染色体8q23.3,全长260 505 bp,包含7个外显子,编码1个由9个锌指结构域组成的转录因子[9]。其中唯一1个GATA锌指结构和2个C末端锌指纹蛋白样锌指结构,是TRPS1行使转录功能的关键,对TRPS1蛋白功能有重要作用。锌指结构中包含的核定位序列的丢失,使TRPS1蛋白无法发挥其作为转录因子的功能,从而导致了TRPS的发生[10]。在患者的受累器官中发现TRPS1基因高度表达,包括软骨、发育中的关节、毛囊和鼻腔区域[11],有学者研究认为这可能与TRPS1在胚胎发育时期的高度表达及调节胚胎发育进程和组织细胞分化有关[12]。
根据患儿病情严重程度及检测的基因型,TRPS共分为三种类型:TRPSⅠ型、TRPSⅡ型和TRPSⅢ型。TRPSⅠ型(MIM 190350)以颅面和骨骼异常为特征,可伴有身材矮小,通常因TRPS1基因无义突变导致,MOMENI等[13]研究表明由特定锌指蛋白(TRPS1)的单倍不足引起此病发生。TRPSⅡ型(MIM 150230)又称Langer-Giedon综合征,由8q24处包含EXT1(外生性骨软骨瘤基因)和TRPS1基因的微缺失引起,该综合征结合了两种常染色体显性遗传疾病的临床特征,即TRPSⅠ型和Ⅰ型遗传性多发性骨软骨瘤(MIM 133700)[14],TRPSⅡ型的特征是除颅面和骨骼的异常外还常伴多发性、外生性骨软骨瘤及智力发育迟缓。TRPSⅢ型(MIM 190351)被认为是Ⅰ型病情更重中的亚型,是由于TRPS1基因无义突变可以减少功能性TRPS1基因拷贝的数量(即单倍体不足),从而导致TRPSⅠ表型。相比之下,GATA型基因序列中的错义突变对转录调控产生显性负效应,从而导致更严重的TRPSⅢ型,通常表现为更严重的骨骼异常,以显著的身材矮小和短指(趾)为特点[15]。
本例患儿根据ACMG指南[7]确诊为TRPS,因其没有智力迟钝、外生骨疣,二代测序未发现EXT1基因,不考虑TRPSⅡ型,考虑Ⅰ型和Ⅲ型。查阅文献,Ⅰ型和Ⅲ型表现相似,主要以临床症状区分,Ⅲ型临床表现更重,通常表现为更严重的骨骼异常及明显的手足短缩畸形[15]。本例患儿就诊时身高95 cm,位于P3百分位,无显著性矮小及骨骼畸形,未见明显的手足短缩,考虑TRPSⅠ型可能性大。此外,当TRPS患者临床特征不典型时,还可以借助分子基因检测协助诊断。
本例患儿因生长发育迟缓、特殊面容就诊,经二代测序发现其TRPS1_ex6 c.2725dupT(p.Cys909Leufs*42)发生杂合移码突变,导致第909位氨基酸由半胱氨酸替换为亮氨酸并改变阅读框,在下游第41位密码子处产生终止密码子,导致翻译提前终止。迄今为止,TRPS1基因现已发现50多个突变,包括缺失、无义和错义突变等。检索国内外数据库,未发现此位点突变的报道,该位点突变为新发变异。本例患儿父母也进行了基因检测,TRPS1基因位点无此变异,该患儿为杂合变异,建议对此类患儿家庭成员的基因进行遗传诊断检测,并提供计划生育咨询有助于进一步分析复发风险[16]。
生长激素-胰岛素样生长因子1(GH-IGF-1)轴在儿童生长发育中起着关键作用[17],IGF-1主要受GH调节,随GH分泌状态发生变化,GHD患儿常表现为GH-IGF-1轴功能紊乱[18]。国外学者MERJANEH等[19]在一项GH-IGF-1轴正常的TRPSⅠ型患儿的研究中使用rhGH治疗,身高在2年内增加了1 SD,与同一时期未经治疗的兄弟姐妹相比,加快了生长速度。提示尽管GH-IGF-1轴正常,GH仍可以治疗TRPSⅠ型患儿,改善其身高。2020年李利等[20]报道了1例GH-IGF-1轴正常的TRPSⅠ型患儿,但是未使用rhGH治疗。rhGH治疗可能改善TRPS儿童生长速度的机制尚未阐明。在模拟TRPS1突变的细胞培养模型中,通过用microRNA阻断正常的TRPS1表达降低了源自鼠畸胎瘤(ATDC5)的软骨形成细胞系的IGF-1表达[21]。因此,在rhGH治疗中产生的全身高浓度IGF-1,可能补偿具有TRPS1突变的个体的生长板中的局部低浓度IGF-1。本报告中,该患儿起始治疗前低水平的IGF-1浓度在rhGH治疗期间也明显升高。
本例患儿根据2008年矮身材儿童诊治指南[8]确诊为GHD,因此本患儿明确诊断为TRPSⅠ型合并GHD。尽管身材矮小是TRPS患者的常见特征,但关于rhGH治疗TRPS合并GHD患者的报告却很少,截至2022年4月,国内未见相关报道。本例患儿rhGH治疗剂量为0.100 U·kg-1·d-1皮下注射治疗,治疗14个月,身高增长12 cm,年身高增长10.3 cm,与2020年以色列报道[4]的1例TRPSⅠ型合并严重GHD患儿,使用rhGH治疗后第一年身高增长9 cm治疗效果相仿。而2012年韩国报道[5]的2例TRPSⅠ型合并GHD患儿使用rhGH治疗结果显示,1例女性患儿身高增长不明显,另1例男性患儿身高无增长,分析该女性患儿疗效不佳的原因可能跟低GH剂量(每周0.2 mg/kg)、遗传身高偏矮(157±5)cm以及患儿依从性较差有关;而男童治疗欠佳可能跟起始治疗年龄较大(14岁),骨骺接近闭合有关。因此,在TRPSⅠ型合并GHD患儿的治疗中,建议使用足量rhGH,并在青春期之前开始治疗,同时加强治疗的依从性,以获得良好的治疗效果。本报道中患儿身高增长明显,治疗效果良好可能与以下因素有关:患儿确诊年龄为3岁9个月,遗传身高为(171±5)cm,确诊后即开始使用rhGH治疗,治疗过程中依从性好,基本无漏针现象,能配合饮食、睡眠,进行足量的纵向运动等。
综上所述,本文报道1例确诊为TRPS合并GHD的患儿,经过基因检测显示其TRPS1_ex6 c.2725dupT(p.Cys909Leufs*42)杂合移码突变为新发变异,给予rhGH治疗1年2个月后,身高为107 cm,HtSDS为-1 SD,身高增长12 cm,年身高增长10.3 cm。观察结果支持评估TRPS和身材矮小患儿的GH-IGF-1轴,并证明rhGH在短时间内可改善TRPSⅠ型合并GHD患儿的身高,且未引起患儿颅内压增高、甲状腺功能减退等不良反应,但对最终成年身高的影响,仍然需要对该患儿进行更长时间的随访,直至成年。
本文诊疗提示:本例患儿心脏彩超提示房间隔卵圆孔未闭,宽约0.34 cm,TRPS通常会合并其他系统的临床表现,如内分泌紊乱、肾脏改变、膀胱输尿管反流和心脏发育异常等,国内也有报道1例房间隔缺损的TRPS男婴[22],并且伴随多系统疾病。因此,需加强对该病的认识,密切随访,以免临床误诊和漏诊。目前TRPS尚无有效治疗方法,临床多以对症处理为主,当身材矮小且生长速度低下,可考虑使用rhGH治疗;当幼年变形性骨软骨炎样股骨头变形需骨科随访,必要时手术治疗;当发现多发性、外生性骨软骨瘤时,可行手术切除等。
陈波,刘青.毛发鼻指(趾)骨综合征合并生长激素缺乏症一例并文献复习[J].中国全科医学,2023,26(21):2686-2689,2694. DOI:10.12114/j.issn.1007-9572.2022.0414. [www.chinagp.net]
CHEN B,LIU Q. Tricho-rhino-phalangeal syndrome with growth hormone deficiency:a case report and literature review[J]. Chinese General Practice,2023,26(21):2686-2689,2694.
本文无利益冲突。

























