
家族性肺纤维化(FPF)定义为家族中有2个或2个以上成员出现肺间质纤维化,为隐匿性遗传病。约半数的FPF患者在第2次就诊时发现,另有半数病例直到家系内出现第2例患者才得以诊断,看似散发性的特发性肺纤维化,而实质上为隐匿性遗传病家系。基因测序是通过血液等体液或细胞对DNA进行测序的技术,可用于FPF的诊断及风险预估。研究报道超过20%的特发性肺纤维化患者有家族史,但目前中国对于肺纤维化患者进行基因检测及家系遗传分析的重视程度较低。中国尚无大规模的FPF统计,以病例报道为主。因此,为了提高对此病的认识及重视,本文对FPF研究进展进行综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)是最常见的特发性间质性肺炎,好发于老年人,男性多于女性,主要症状和体征包括呼吸困难、咳嗽、Velcro啰音和杵状指[1]。研究报道超过20%的IPF患者有家族史,诊断为家族性肺纤维化(familial pulmonary fibrosis,FPF)[2]。其定义为家族中有2个或2个以上成员出现肺间质纤维化,为罕见病[1]。目前FPF发病机制不明,且临床资料较少。阐明FPF的发病机制,可进一步指导优生优育及阻断疾病的发生。本文将从FPF的定义、遗传机制及相关基因、临床表型特点、治疗及预后方面进行阐述。
2000年,Marshall等[3]报道了英国25个FPF家族,共67例FPF患者,男女比例为1.75∶1,平均诊断年龄为55.5岁。Lee等[4]在1992年到2002年间筛选了15个FPF家族,共47例FPF患者,男女比例为2∶1,其中27例FPF患者的平均发病年龄和诊断年龄分别为58.3岁和59.4岁。芬兰FPF的患病率约为5.9/100万[5],英国FPF的患病率约为1.34/100万[3]。我国目前尚无较大规模的FPF统计,以病例报道为主,共报道7个家族共22例肺纤维化(表1)。
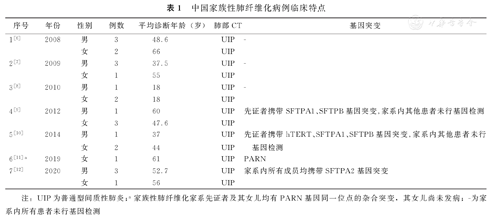
中国家族性肺纤维化病例临床特点
中国家族性肺纤维化病例临床特点
| 序号 | 年份 | 性别 | 例数 | 平均诊断年龄(岁) | 肺部CT | 基因突变 |
|---|---|---|---|---|---|---|
| 1[6] | 2008 | 男 | 3 | 48.6 | UIP | - |
| 女 | 2 | 66 | UIP | |||
| 2[7] | 2009 | 男 | 3 | 37.5 | UIP | - |
| 女 | 1 | 55 | UIP | |||
| 3[8] | 2010 | 男 | 1 | 18 | UIP | - |
| 女 | 2 | 18 | UIP | |||
| 4[9] | 2012 | 男 | 1 | 60 | UIP | 先证者携带SFTPA1、SFTPB基因突变,家系内其他患者未行基因检测 |
| 女 | 3 | 47.6 | UIP | |||
| 5[10] | 2014 | 男 | 1 | 37 | UIP | 先证者携带hTERT、SFTPA1、SFTPB基因突变,家系内其他患者未行基因检测 |
| 女 | 2 | 44 | UIP | |||
| 6[11]a | 2019 | 女 | 1 | 61 | UIP | PARN |
| 7[12] | 2020 | 男 | 3 | 52.7 | UIP | 家系内所有成员均携带SFTPA2基因突变 |
| 女 | 1 | 56 | UIP |
注:UIP为普通型间质性肺炎;a家族性肺纤维化家系先证者及其女儿均有PARN基因同一位点的杂合突变,其女儿尚未发病;-为家系内所有患者未行基因检测
研究报道FPF有多种遗传方式,多数倾向于外显率可变的常染色体显性遗传[1,4],目前仅有1例常染色体隐性遗传的报道[13]。近年来,FPF遗传基因方面的研究报道越来越多,FPF的遗传机制可能参与了散发性肺间质疾病尤其是IPF的发病机制。目前20%的致病基因已经明确,另有80%尚未明确致病基因[14]。已报道的与FPF相关的主要基因如下所述。
Ⅱ型肺泡上皮细胞能够合成分泌大量肺表面活性物质,包含脂类和蛋白质。肺表面活性物质中8%~10%的蛋白质能与脂质特异性结合,定义为肺表面活性物质相关蛋白(surfactant proteins,SPs)。目前已发现的SPs有4种,分别为SP-A、SP-B、SP-C、SP-D,相应调控基因分别为SFTPA、SFTPB、SFTPC、SFTPD。
2001年,Nogee等[15]首次在FPF家系中发现了SFTPC杂合突变,即SFTPC内含子剪接区4号位点的核苷酸G被A取代,使编码SP-C前体蛋白(pro-SP-C)的4号外显子漏读,导致编码蛋白缺失37个氨基酸,SP-C缺乏。随后文献陆续报道了SFTPC 40多个突变位点。SFTPC基因突变多见于FPF,散发IPF中少见,流行率低于1%[16]。SFTPC编码pro-SP-C,pro-SP-C水解数次后裂解为SP-C分泌至肺泡腔。pro-SP-C的C末端定位于内质网腔内,含有BRICHOS结构域。BRICHOS结构域对pro-SP-C起伴侣作用,对pro-SP-C的成熟、正确折叠及包装至关重要[17]。FPF的SFTPC突变多发生于BRICHOS结构域的编码区,突变后SP-C蛋白折叠异常或成熟障碍,诱发内质网应激,抑制泛素/蛋白酶体系统,最终导致肺纤维化,促进细胞凋亡[18,19]。
SFTPA1和SFTPA2位于10号染色体长臂,共同编码SP-A。2009年,Wang等[20]通过对1个FPF家系进行连锁共分离分析,在FPF和肺腺癌患者中发现了SFTPA2等位基因231位密码子的颠换突变(GGG to GTG)。2016年,Nathan等[21]报道了一个FPF和肺癌的家系,发现SFTPA1第631位点的核苷酸G被T取代,导致编码蛋白的211位色氨酸残基被精氨酸取代。SP-A是一种与凝集素同源的多聚体蛋白,在先天防御系统和肺的免疫应答中发挥作用。SFTPA突变激活非折叠蛋白反应,促进编码免疫球蛋白结合蛋白报道基因的转录,免疫球蛋白结合蛋白表达增加,内质网应激生成X盒结合蛋白1剪接物,最终加速肺泡上皮细胞凋亡[22]。
ABCA3主要表达于Ⅱ型肺泡上皮细胞,为细胞膜表面ATP结合区的脂质转运蛋白,参与肺表面活性物质的加工和运输。Zhou等[23]报道ABCA3基因突变可能与中国人群的肺间质疾病易感性增加相关。
端粒是DNA末端一段多鸟嘌呤的重复序列,保护染色体和基因的完整性。端粒随细胞分裂而损耗,缩短到一定长度后,细胞便无法启动分裂,走向衰老和凋亡。端粒酶存在于干细胞、生殖细胞以及恶性肿瘤细胞等细胞中,通过在染色体末端添加端粒重复序列(TTAGGG)来抵消细胞分裂所致的端粒缩短,由端粒酶RNA、端粒酶逆转录酶(telomerase reverse transcriptase,TERT)和相关蛋白组成。研究报道,肺纤维化是端粒病谱上的主要疾病之一,端粒酶相关基因(如TERT、TERC、RTEL1、PARN、TINF2和DKC1等)突变占FPF病例总数比例约多达25%,在散发性IPF中约占1%~3%[24]。
TERC和TERT分别位于第3号染色体长臂和第5号染色体短臂。2007年,Armanios等[25]在73例FPF先证者中发现了5例TERT突变和1例TERC突变,并在突变患者中检测到端粒缩短。通过对比先证者、携带者和正常人群的端粒长度,发现先证者和携带者的端粒平均长度比正常人群明显缩短,表明携带基因突变的无症状人群也有患FPF的风险。目前关于TERT和TERC突变导致肺纤维化的具体机制尚不明了。有研究表明,Ⅱ型肺泡上皮细胞端粒功能障碍可引起DNA损伤应答,激活p53通路,从而诱导细胞衰老及肺上皮细胞稳态失衡[26]。TERT和TERC突变可能激活Wnt-β-catenin通路,升高TGF-β,促进上皮间质转换,但仍需进一步研究证实[27]。
DKC1基因编码端粒酶复合物成分,参与核糖体合成和端粒修复。DKC1突变可导致先天性角化不良,超过20%的患者可并发肺部疾病[28]。2014年,Kropski等[29]报道了1例FPF家系先证者的DKC1新位点突变(c.1213A>G)。目前关于DKC1致肺纤维化的机制尚未明了,仍需进一步探索。
2015年,Stuart等[30]首次报道了2个与端粒酶长度相关的新基因(PARN和RTEL1)与FPF有关。随后Kropski等[31]在188个FPF家系中发现13个家族有PARN突变。2018年,中国也报道了1例FPF家系PARN基因突变[12]。RTEL1参与端粒的修复,是先天性角化不良的突变位点之一,FPF患者比对照组的RTeL1基因保守位点的突变数目更多,有害程度更高(P=1.6×10-6)[30]。PARN和RTEL1基因参与肺纤维化的机制仍需进一步研究。
MUC5B是气道分泌性黏蛋白的成分之一,主要由黏膜下腺合成,正常主要分布在传导性支气管,在气道黏液纤毛清除、调节气道炎症、防御肺部感染方面发挥重要保护作用。MUC5B基因位于染色体11p15的保守簇,主要调控气道黏蛋白MUC5B合成。MUC5B是IPF和FPF的等位危险基因,研究发现位于MUC5B启动子的SNP(rs35705950)与FPF和IPF相关性最强,存在于34%的家族性肺间质病和38%的IPF,仅有9%存在于正常对照[32]。文献报道MUC5B rs35705950启动子G>T多态性与中国男性IPF患者遗传易感性相关[33]。MUC5B基因突变导致MUC5B分泌过多,潴留在肺泡和蜂窝组织内,反而导致肺清除能力下降,随着时间的推移,肺慢性损伤最终发展为肺纤维化。Chen等[34]报道内质网核蛋白2触发X盒结合蛋白1剪接,剪接的X盒结合蛋白1通过MUC5B启动子变异依赖途径上调气道上皮细胞中MUC5B的表达,所以抑制内质网核蛋白2依赖通路/元件可能为IPF的治疗提供新途径。MUC5B在病理情况下受到肺微环境及疾病发生的相互影响异常表达,其参与肺纤维化发病的具体机制有待进一步研究证实。
FPF诊断主要依赖于高分辨率CT影像表现符合肺纤维化及结合家族史,必要时可行肺穿刺活检明确诊断[3]。FPF患者通常起病隐匿,发病年龄较早,男性多于女性,主要症状和体征与IPF患者相似,表现为干咳、进行性劳力性呼吸困难等,多数患者双肺下部可闻及吸气相Velcro啰音,部分患者可出现杵状指(趾)。晚期可出现肺动脉高压、肺源性心脏病等并发症[3,4]。
FPF的高分辨率CT主要表现为普通型间质性肺炎,即伴或不伴外周牵引的牵拉性支气管扩张或支气管扩张的蜂窝影,以胸膜下肺基底部分布为主,分布往往具有异质性。少部分表现为非特异性间质性肺炎、隐源性机化性肺炎、过敏性肺炎、肺中央结节以及未分类的间质性肺炎[35]。
FPF的病理学表现主要为普通型间质性肺炎,即明显肺纤维化伴结构扭曲(破坏性瘢痕伴或不伴蜂窝样改变),纤维化主要分布在胸膜下和/或间隔旁;肺实质有斑片状纤维化累及;成纤维细胞灶。部分表现为非特异性间质性肺炎、隐源性机化性肺炎、过敏性肺炎以及未分类的间质性肺炎等[35]。
FPF的肺功能表现为限制性通气功能障碍和弥散功能降低,肺活量、肺总量减少,功能残气量和残气量降低,第1秒用力呼气容积与FVC之比正常或增加,流速容量曲线的最大峰值增加,通气血流比例失衡。
风湿因子、自身免疫性抗体等血清学检查主要用于排除继发性因素所致的肺纤维化,如自身免疫性疾病肺受累等。建议进行自身抗体谱测定以及血尿常规、肝肾功能、肌酶等测定,用于判断有无多脏器受累等,均有助于鉴别诊断。
FPF预后欠佳,发病后平均中位生存期仅7年左右[36]。但因FPF为家族遗传性疾病,目前治疗上尚无明确有效的方法。Koga等[37]报道了1例通过吡非尼酮治疗后肺纤维化程度及呼吸困难症状明显好转的FPF病例。最近有文献报道FPF和IPF对吡非尼酮耐受性较好,两者使用吡非尼酮的不良反应差异无统计学意义,但仍需大样本临床试验进一步证实[38]。目前尚无文献报道肺移植对于FPF的治疗效果,仍需进一步研究。
FPF尚无有效的药物治疗,病死率高,但由于其在临床上罕见,早期发现和干预仍较为有意义。对于有家族史的肺纤维化患者,其无症状的家庭成员应加强监测,避免吸烟等可能会导致肺纤维化的危险因素,并进行早期的遗传学咨询。
所有作者均声明不存在利益冲突

























