
慢性阻塞性肺疾病(COPD)的发生、发展过程中,多种免疫细胞发挥了重要作用,巨噬细胞是其中之一。肺泡巨噬细胞(AM)作为肺部免疫细胞的重要组成部分,存在于肺泡微环境中,其表型、功能和表型转化等均受到肺泡微环境的影响。AM既可作为促炎者(M1)释放炎症因子、氧化剂和蛋白酶,也可作为抗炎者(M2)吞噬和清除病原微生物和凋亡细胞碎片。了解COPD中AM的变化并将其作为干预或者治疗靶点或许是COPD治疗的新思路。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
COPD的特征是存在呼吸道和(或)肺泡异常导致的持续的呼吸道症状和气流受限,这通常是由大量接触有毒气体和颗粒物引起,吸烟是COPD的主要危险因素,COPD的急性加重期和合并症常影响患者的质量调整生命年[1]。COPD目前被列为第四大死因,全世界共有3.28亿例确诊患者[2]。王辰院士团队2018年完成的我国COPD发病率和危险因素横断面研究显示,我国20岁及以上人群中COPD的发病率约为8.6%,据此推算我国20岁及以上COPD患者数量约为1亿人;此外,大部分COPD患者对自身病情不知情,诊断率远低于发病率,这导致在就医时,其第1秒用力呼气容积(forced expiratory volume in the first second,FEV1)/FVC至少低于60%,已延误最佳治疗时机;早发现、早诊断、早干预是我国COPD防治的主要任务[3]。
人体呼吸道是重要的免疫界面,肺部微环境中存在大量共生菌群及固有免疫细胞,在肺动态免疫平衡中起主要作用。巨噬细胞是固有免疫的重要成员,在COPD发病过程中起关键作用,其通过抗原呈递,对细菌、细颗粒物、凋亡细胞等多种抗原识别、吞噬和清除,参与炎性反应的发生、发展及机体的免疫防御调节。肺部微环境中至少存在3种巨噬细胞:支气管巨噬细胞、肺间质巨噬细胞和肺泡巨噬细胞(alveolar macrophages,AM)。AM是远端肺组织中数量最多、功能最重要的部分,其既可来源于肺间质巨噬细胞,也可来源于循环血单核细胞,在COPD患者的支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)和痰液中,AM数量明显增加。目前对于COPD发病机制的认识,主要有氧化应激、炎性反应、蛋白酶-抗蛋白酶失衡、细胞凋亡等,AM在其中均有参与,是COPD发病过程中的核心成员之一[4]。
COPD患者中超氧化物歧化酶、过氧化氢酶、核因子-E2相关因子2(nuclear factor erythroid derived 2-like 2,Nrf2)、组蛋白去乙酰化酶2活力降低,还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶、黄嘌呤氧化酶、血红素过氧化物酶、丙二醛、8-羟基脱氧鸟苷表达增多;利用液相色谱质谱联用技术分析香烟烟雾在COPD患者体内的代谢,发现其体内的氧化应激水平显著高于对照组,且COPD患者氧化应激水平与肺功能呈负相关,提示氧化/抗氧化失衡与COPD发病密切相关[5,6]。
肺部氧化应激中的氧化剂分为内外两部分,内源性氧化剂主要由AM等多种细胞分泌,外部氧化剂则源自香烟烟雾及PM2.5等细颗粒物。外部氧化剂可增加体内氧化负担,刺激中性粒细胞和巨噬细胞释放内源性活性氧;活性氧可抑制磷酸酶与张力蛋白同源物从而激活磷脂酰肌醇3-激酶,导致雷帕霉素靶蛋白(mechanistic target of rapamycin,mTOR)表达增加,mTOR可激活p70核糖体蛋白S6激酶、microRNAs-34a和microRNAs-570,从而抑制沉默信息调节因子1,导致细胞衰老和线粒体功能障碍,释放线粒体活性氧。尽管COPD患者体内AM基础线粒体活性氧释放增加,但在肺炎链球菌激发后其线粒体活性氧生成不增加,从而减弱AM的杀细菌作用,这一过程与髓系白血病细胞分化蛋白1表达增加相关[7](图1)。COPD病程中AM诱导型一氧化氮合酶、活性氮及过氧亚硝酸盐阴离子、转化生长因子β(transforming growth factor-β,TGF-β)表达均增多;转录因子叉头框蛋白活性则降低,抑制超氧化物歧化酶及过氧化氢酶的表达。活性氧还可激活核因子-κB(nuclear factor-κB,NF-κB)、激活蛋白1等胞内信号通路,上调炎症介质的表达,放大炎症效应;激活RAS同源基因家族成员A促进巨噬细胞重构,破坏甘露醇结合凝集素,损伤巨噬细胞的吞噬功能,从而影响肺部免疫平衡与凋亡细胞清除[8]。
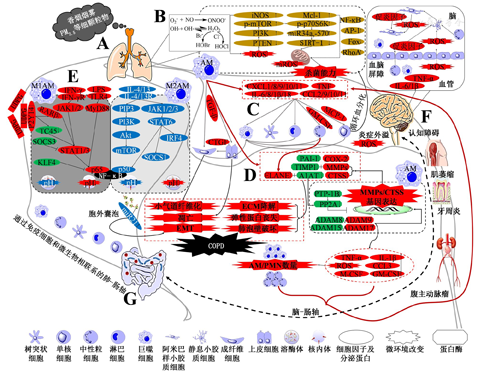
注:iNOS为诱导型一氧化氮合酶;p-mTOR为磷酸化雷帕霉素靶蛋白;PI3K为磷脂酰肌醇-3-激酶;PTEN为磷酸酶与张力蛋白同源物;ROS为活性氧;Mcl-1为髓系白血病细胞分化蛋白-1;p-p70S6K为磷酸化p70核糖体蛋白S6激酶;SIRT-1为sirtuin-1;mROS为线粒体活性氧;NF-κB为核因子-κB;AP-1为激活蛋白-1;Fox为转录因子叉头框蛋白;RhoA为RAS同源基因家族成员A;TNF-α为肿瘤坏死因子α;CXCL为趋化因子CXC样配体;CCL为趋化因子CC样配体;MCP-1为单核细胞趋化蛋白-1;GM-CSF为粒细胞-巨噬细胞集落刺激因子;TGF-β为转化生长因子β;CTGF为结缔组织生长因子;ELANE为弹性蛋白酶;PAI-1为纤溶酶原激活物抑制剂-1;TIMP为金属蛋白酶组织抑制物;A1AT为α1-抗胰蛋白酶;COX-2为环氧合酶-2;MMPs为基质金属蛋白酶;CTSS为组织蛋白酶S;PTP-1B为蛋白酪氨酸磷酸酶1B;PP2A为蛋白磷酸酶2A;ADAM为去整合素和金属蛋白酶;M-CSF为巨噬细胞集落刺激因子;AM为肺泡巨噬细胞;PMN为中性粒细胞;ECM为细胞外基质;EMT为上皮间质转化;IFN-γ为干扰素γ;IFN-γR为干扰素γ受体;JAK为Janus激酶;LPS为脂多糖;TLR4为Toll样受体4;MyD88为髓样分化因子MyD88;RARβ为胞核视黄酸受体β;TC45为T细胞蛋白酪氨酸磷酸酶45 kD同工型;SOCS为细胞因子信号抑制子;STAT为信号转导和转录激活因子;KLF4为红系Kruppel样因子4;pH为酸碱度;UDPG为二磷酸尿苷葡糖;P2Y14为嘌呤能G蛋白耦联受体14;PIP3为三磷酸(3,4,5)磷脂肌醇;Akt为Akt丝氨酸/苏氨酸激酶家族;IRF为干扰素调节因子;miR为微小核糖核酸
此外,体外暴露于香烟烟雾的AM及COPD患者体内AM中存在代谢重编程现象,其代谢途径从线粒体氧化转变为糖酵解,这一现象与炎症因子的升高相关;COPD患者体内AM的线粒体总数和质量均较低,其已经失去了通过增加糖酵解动态补偿线粒体功能障碍的能力,利用特定Nrf2激动剂刺激COPD小鼠AM,除增加Nrf2核转位,调节其代谢途径外,还可抑制其炎症因子释放并调节其吞噬功能[9,10]。
炎症在COPD导致的气道病变中起重要作用,轻、中度患者存在肺动脉壁增厚、炎性细胞浸润及血管松弛失调等症状;其血清中C反应蛋白、内皮素1、脑钠肽、肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)、IL-18等炎症标志物以及BALF和痰液中IL-6、IL-8、IL-10、IL-1β、TNF-α等炎症标志物均高于健康人群[11]。利用转录组分析人源体外气道组织模型在香烟烟雾暴露中的病理过程,发现白介素1受体拮抗剂、IL-1β、IL-6、IL-8、IL-12、血管内皮生长因子等细胞因子表达增多,且存在香烟烟雾剂量依赖性[12]。
炎症反应与多种细胞相关,AM、中性粒细胞、肥大细胞、树突状细胞、单核细胞、自然杀伤细胞等免疫细胞以及气道、肺泡上皮细胞等均可分泌IL-6、IL-8、IL-10、IL-1β、TNF-α、细胞间黏附分子等多种细胞因子,诱导包括成纤维细胞在内的多种细胞增殖、分化及迁移[13,14]。多种细胞如肺部固有免疫细胞不仅可作为AM活化增殖的上游细胞,也可被AM募集,迁移入肺泡,AM分泌的趋化因子CC样配体(chemokine C-C motif ligand,CCL)2、CCL9、CCL10、CCL11,趋化因子CXC样配体(chemokine C-X-C motif ligand,CXCL)1、CXCL8、CXCL9、CXCL10、CXCL11,及单核细胞趋化蛋白1等可诱导循环单核细胞、巨噬细胞、中性粒细胞、辅助性T淋巴细胞及细胞毒性T淋巴细胞等向肺组织及肺泡迁移,形成炎性浸润,并已在COPD患者的BALF及痰液中得到验证[15]。AM与肺部不同细胞间的"募集"与"被募集"关系形成了复杂的"炎症网络",COPD患者体内AM在香烟烟雾中的反复暴露促进了炎症介质的释放,为炎症网络提供了多个炎症反应的"放大环"(图1),循环血单核细胞向AM的分化则驱动着炎症网络的持续运行,可以说,AM在COPD炎症网络中起关键的中枢作用[16]。
除肺部慢性炎症外,COPD患者普遍存在全身性炎症,主要表现在COPD患者并发牙周炎、骨密度降低、肌肉萎缩及腹主动脉瘤等疾病,一个更严重表现是部分COPD稳定期患者并发认知障碍,其潜在机制为炎症外溢促使体循环中的炎症因子、趋化因子和免疫细胞水平升高,破坏血脑屏障内皮细胞间的紧密连接,增加血脑屏障通透性或产生缺乏有效血脑屏障的区域,引发中枢性炎症,导致白细胞渗入大脑,小胶质细胞激活和增殖,NF-κB激活,炎症因子的激活和高表达,以及活性氧产生[1,6,17](图1)。
AM可分泌多种蛋白酶,以基质金属蛋白酶(matrix metalloproteinases,MMPs)为主,MMPs可被金属蛋白酶组织抑制物(tissue inhibitors of metalloproteinases,TIMPs)特异性内源性抑制。COPD临床研究及COPD动物模型中均发现肺组织及BALF中MMP-1、MMP-2、MMP-3、MMP-7、MMP-8、MMP-9、MMP-12、MMP-14转录增多,活性增强,而TIMP-1表达降低,其中MMP-2及MMP-9主要由AM分泌,且MMP-9浓度与FEV1呈负相关,MMP-9/TIMP-1比值与气流阻塞程度呈负相关;MMPs与上皮间质转化相关,MMP-3、MMP-7可减少细胞间附着,MMP-2、MMP-9则参与基底膜的破坏,导致细胞分离和细胞运动性增强;MMPs还引起细胞外基质的降解和沉积失衡,其与环氧合酶2相协同,与纤溶酶原激活物抑制剂1相拮抗。MMPs还可介导炎症因子的释放,如TNF-α可经MMP-12途径释放,提示MMPs参与炎症网络;MMPs作用产生的纤维蛋白及层黏连蛋白片段可趋化免疫细胞,增强肺部炎症放大效应[18]。
AM与中性粒细胞还可分泌弹性蛋白酶,其可降解弹性蛋白并促进肺泡上皮细胞凋亡,其内源性抑制剂为α1-抗胰蛋白酶(α1-antitrypsin,A1AT)。生理状态下,组织内90%的弹性蛋白酶可与A1AT结合失活,病理状态下大量聚集的AM和中性粒细胞释放的弹性蛋白酶难以被完全灭活;内源性活性氧氧化A1AT的甲硫氨酸残基,使A1AT失活,造成弹性蛋白酶/A1AT失调,临床研究证明A1AT可以减少肺内弹性蛋白的降解,减轻气道炎症[19]。具有功能活性的A1AT蛋白与蛋白酪氨酸磷酸酶1B共同作用激活AM、单核细胞、中性粒细胞、气道上皮细胞中的蛋白磷酸酶2A(protein phosphatase 2A,PP2A)。PP2A可减少香烟烟雾诱导的组织蛋白酶S,组织蛋白酶S基因敲除小鼠可抵抗香烟烟雾急性暴露引起的炎症、气道高反应性、肺大泡形成和肺功能丧失;PP2A缺失可增强肺损伤和肺纤维化,并导致IL-10依赖的上皮细胞凋亡敏感性增强(图1);通过调控PP2A,A1AT可抑制TNF-α对MMPs、炎症因子的诱导作用[20,21]。
去整合素和金属蛋白酶(a disintegrin and metalloprotease,ADAM)在COPD中的作用也值得关注,AM中ADAM9的表达可增加AM诱导MMPs、氧化剂、炎症因子等产生的肺负荷,造成细胞外基质、弹性蛋白降解以及肺泡壁的破坏;ADAM17在因缺氧导致的阻塞性支气管哮喘进展为肺纤维化中可见高表达;ADAM8、ADAM15则具有保护作用,AM中ADAM8表达缺乏可导致AM数量增多,伴随氧化应激增加和炎症因子释放增多,类似现象可在AM中ADAM15表达缺乏时产生[22,23,24,25](图1)。
AM激活主要有两种表型,即起促炎作用的经典激活途径M1型及起抗炎作用的替代激活途径M2型,且两者间动态可逆,主要依靠细胞表面标志物、分泌不同细胞因子及趋化因子进行区分,通过干扰素γ受体-Janus激酶1/2-信号转导和转录激活因子(signal transducers and activation of transcription,STAT)-1/3-NF-κB p65轴及Toll样受体4-髓样分化因子MyD88-NF-κB p65轴可诱导AM向M1型极化;M2型极化则由IL-4R/13R-Janus激酶1/2/3-STAT6-细胞因子信号抑制子1/干扰素调节因子4/NF-κB p50轴及IL-4R/13R-三磷酸(3,4,5)磷脂肌醇-磷脂酰肌醇3-激酶-丝氨酸/苏氨酸激酶Akt1-mTOR轴调控[26](图1)。COPD病程中存在M1、M2表面标志物同时表达的过渡态,M1标志物表达随疾病进展减少,M2标志物表达则持续升高,这提示M1/M2失衡或与COPD进展相关,其潜在机制为M1极化产生的促炎介质参与炎症网络的发生、发展;当M1/M2平衡发生倾斜时机体为维持稳态增加M2极化;M2极化产生在蛋白酶-抗蛋白酶失衡中起关键作用的MMP-12,并通过TGF-β/SMADs轴减少炎症因子表达;M2极化同时促进COPD中上皮间质转化的产生及发展,这与胞外囊泡中microRNAs-21水平较高相关,上皮细胞可通过减少胞外囊泡中microRNAs-21含量抑制M2 AM产生;维持M1/M2在肺部微环境中的动态平衡对稳定COPD患者病情,减缓COPD急性加重及逆转早期COPD患者的疾病状态有重要作用[18]。
基于转录组学的网络分析发现,COPD患者与吸烟者相比,体内糖皮质激素相关基因模块耗竭更显著。此外,COPD患者AM的主要特征为抗原处理、炎症应答和免疫应答功能的丧失[27]。AM极化具有高度可塑性,这在SARS-CoV-2感染过程中也有体现,M1 AM与M2 AM相比,其核内体pH值更低,溶酶体pH值更高;病毒DNA/RNA可作为病原体相关分子模式促进M2 AM向M1 AM转变,炎症因子也可促进此过程(图1)[28]。作为巨噬细胞M1极化的经典刺激剂,脂多糖/干扰素γ可刺激巨噬细胞合成糖原,通过糖原分解产生葡萄糖-6-磷酸,并通过磷酸戊糖途径产生还原型烟酰胺腺嘌呤二核苷酸磷酸,确保M1巨噬细胞生存所需的还原型谷胱甘肽;糖原代谢可上调UDPG/P2Y14轴表达,上调RARβ以促进STAT1表达,下调T细胞蛋白酪氨酸磷酸酶45以促进STAT1磷酸化,提示应关注能量代谢对AM极化的影响[29]。CD206、CD163等M2表面标志物与COPD患者肺部巨噬细胞吞噬能力相关,且肺间质巨噬细胞与AM相比,其吞噬性能较差,可以说,M2 AM在吞噬肺部凋亡细胞中发挥关键作用;由于COPD病程中存在下呼吸道细菌定植,尽管COPD后期M2 AM数量上升,其仍不足以吞噬清除细颗粒物及M1 AM杀微生物作用遗留的细胞碎片,表现为AM吞噬功能的减弱[18]。鼻病毒感染是COPD急性加重的主要原因之一,其可诱导AM减少CXCL8表达或IL-1R相关激酶的降解,降低细菌清除率,诱导AM细菌耐受[30]。气道黏膜微环境对AM存在表观遗传重编程作用,可影响AM表型,如黏液阻塞性肺疾病小鼠模型中,黏液暴露会损害AM的胞葬作用和吞噬能力[31];此外,香烟烟雾暴露会增加AM、中性粒细胞等的自噬,且自噬水平与吞噬能力呈负相关,自噬抑制剂3-甲基腺嘌呤可抑制香烟烟雾刺激时巨噬细胞MMP-12的表达,并抑制香烟烟雾暴露导致的小鼠支气管炎样表型[32]。
慢性阻塞性肺疾病全球倡议将COPD患者按气流受限严重程度分为Ⅰ~Ⅳ级,其肺组织中髓样细胞触发受体2(triggering receptors expressed on myeloid cells 2,TREM2) mRNA水平升高,且与CD68、CD206及几丁质酶共表达,提示TREM2或可反映COPD患者AM M2极化状态,此外,TREM2/TREM1 mRNA水平比值的升高与FEV1及FEV1/FVC的降低相关[33]。粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colony-stimulating factor,GM-CSF)对于AM的吞噬功能十分重要,缺乏GM-CSF会导致肺表面活性蛋白在气道内的积聚,从而通过肺表面活性蛋白A/D-含Src同源2结构域蛋白酪氨酸磷酸酶(Src homology-2 domain-containing protein tyrosine phosphatase,SHP)1-RAS同源基因家族成员A轴抑制AM的吞噬功能;利用GM-CSF诱导多能干细胞分化成为肺泡样巨噬细胞,通过气道递送,可在急性肺损伤期间吞噬中性粒细胞,促进肺组织修复,该方法在急性肺损伤小鼠模型中得到验证[34]。混交激酶域蛋白和受体相互作用激酶3作为细胞程序性坏死的驱动蛋白,在重度COPD患者支气管上皮细胞和巨噬细胞中表达均增加,患者肺组织中还检测出高水平的磷酸化的混交激酶域蛋白和受体相互作用激酶3,在香烟烟雾暴露的小鼠COPD模型中也观察到类似现象[35]。
SHP2在远端肺中高表达,AM中SHP2缺失可促进其M2极化,并引起TGF-β失调,造成肺组织中MMP-12持续增加,提示应关注COPD患者肺部SHP2表达量与活性;此外,高表达细胞表面GPR78的AM也可大量释放MMP-12,加速肺部炎症反应[36,37]。利用脂质组学筛选COPD病程中脂类生物标志物,其中前列腺素E2、白三烯、三己糖神经酰胺等含量增加,鞘磷脂含量减少,这种变化与囊性纤维化小鼠模型中AM的变化相类似,会导致慢性炎症的发生与上皮细胞的死亡,与鞘磷脂酶及神经酰胺酶活性改变相关,可通过吸入鞘磷脂酶抑制剂或重组酸性神经酰胺酶纠正[38,39]。肠道微环境与肺部微环境可通过免疫细胞相互影响,即肠-肺轴(图1),通过对口腔、鼻腔和BALF中的微生物区系进行冗余分析发现,BALF与口腔的微生物群落更接近;将健康动物的粪菌移植给受试动物,可恢复受试动物肺部AM及中性粒细胞细菌清除能力和部分细胞因子水平;此外,因老龄化导致的肺部病变也被认为可能与肠道微环境的改变相关[40]。
目前COPD的治疗选择主要为支气管舒张剂,包括长效β2肾上腺素受体激动剂和长效胆碱能受体拮抗剂,其均不能在不引发严重不良反应的情况下减弱COPD的氧化应激和炎症反应;在长效β2肾上腺素受体激动剂和长效胆碱能受体拮抗剂均无效情况下建议使用吸入性糖皮质激素(inhaled corticosteroids,ICS)应对COPD急性加重,但其对于COPD稳定期患者治疗益处不大,且存在多种不良反应[41]。
RV568是专为抗p38-丝裂原激活蛋白激酶的α和γ亚型设计的窄谱激酶抑制剂,其对巨噬细胞分泌CXCL8的抑制作用强于ICS;并可降低香烟烟雾暴露小鼠体内中性粒细胞和巨噬细胞数量,抑制细胞因子释放;RV568联用ICS可产生更强的抑制炎症因子释放作用,提示二者具有协同作用;在COPD患者中,吸入RV568可改善FEV1,降低痰液丙二醛水平[42]。阿胶是含氨基酸、微量元素和胶原蛋白小分子量水解产物的固体胶,其可显著降低PM2.5长期暴露大鼠BALF中的嗜酸粒细胞、淋巴细胞和中性粒细胞数量及TNF-α、IL-1β含量,减少肺部炎症细胞浸润,抑制PM2.5诱导的大鼠肺组织间质/肺泡壁增厚和肺气肿样改变。分析阿胶成分,找到起主要作用的结构明确的单一成分可作为研究方向之一[43]。
Isthmin 1(ISM1)是驻留在肺内的一种抗炎蛋白,其可选择性刺激细胞表面GPR78+ AM凋亡。人肺中ISM1表达量与AM凋亡呈正相关,气道递送重组ISM1及气管给予阿仑膦酸钠和氯膦酸盐均可诱导AM凋亡,改善香烟烟雾诱导的小鼠肺气肿,表明选择性耗竭AM可作为治疗COPD的新策略之一[37]。使用细菌衍生产品也可干预COPD进程,用灭活的克雷伯菌预防性皮下注射给急性香烟烟雾暴露小鼠,可降低香烟烟雾暴露后小鼠BALF中干扰素γ、CXCL9、CXCL10、CCL5、IL-6、粒细胞集落刺激因子和IL-17的表达水平,并减少BALF中巨噬细胞和淋巴细胞的数量,此外这种免疫疗法还诱导了全身免疫的激活[44]。
有抗炎、抗氧化及肺部保护作用的保健品开发及合理应用也可作为预防和保护潜在的COPD患病人群的一种策略。白藜芦醇大量存在于葡萄皮、葡萄籽中,可抑制环氧合酶2转录及环氧合酶1活性,发挥抗氧化作用。白藜芦醇还可通过激活沉默信息调节因子1抑制NF-κB的核移位,减少炎症因子表达,适度补充该类物质在对抗COPD中或可产生积极作用[45]。
AM在维持肺泡免疫微环境稳态中具有重要作用,对其表面受体、相关信号通路、作用机制的了解,将为逆转COPD早期或减缓COPD急性加重期中AM发生的变化提供策略。AM广泛参与COPD的不同发病机制,在其中扮演了重要的角色。以AM为靶标寻找到可以特异性反映COPD不同阶段的生物标志物,以改变AM病理状态为目标找到相关药物或相关途径进行早期干预对延缓COPD进程或者控制急性加重十分重要,值得深入探索。
所有作者声明无利益冲突

























