
肺动脉高压(PH)是一种不同病因及发病机制引起的肺血管结构或者功能的改变,从而引起肺血管阻力及肺动脉压力进行性升高的疾病。PH的发病机制涉及到多个方面,炎症机制在其中扮演着重要的角色。高迁移率族蛋白B1(HMGB1)是一种核蛋白,正常情况下其在细胞核中与DNA结合,参与DNA的复制和修复。而在应激条件下,其释放到胞质及胞外发挥不同的作用。近年来,多项研究证明HMGB1在PH的发生发展中扮演着一定的角色,在实验性PH的治疗中,靶向HMGB1展示了一定的潜能。本文立足于HMGB1,结合其生物学特点,综述HMGB1在不同的细胞位置对PH的发生发展中的作用,为治疗PH提供一条新的思路与途径。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
肺动脉高压(pulmonary hypertension,PH)是一种不同病因及发病机制引起的肺血管结构或者功能的改变,从而引起肺血管阻力及肺动脉压力进行性升高的疾病;其诊断标准为海平面、静息状态下,经右心导管检查测定的肺动脉平均压≥25 mmHg(1 mmHg=0.133 kPa)[1]。PH因肺血管重建和原位血栓形成等因素导致肺内小动脉逐渐增厚的疾病[2],其发病机制与许多分子过程有关,主要包括生殖系突变、炎症、肺动脉内皮细胞功能障碍、表观遗传修饰、DNA损伤、代谢功能障碍、性激素失衡、氧化应激等[3]。很多证据表明,炎症和免疫在PH的发病机制中发挥着关键作用。高迁移率族蛋白B1(high-mobility group box 1,HMGB1)可以通过各种各样的机制促进炎症反应,所以HMGB1持续和过度的释放可能导致肺血管的重构及PH的形成。现就HMGB1与PH发病机制的相关研究综述如下。
HMGB1是一种作为染色质结合因子与DNA结合的核蛋白,参与DNA复制和修复。它于1973年首次从小牛胸腺染色质中提取,并因其在凝胶电泳中的高流动性而命名[4]。HMGB1在结构上由两个DNA结合位点(A盒和B盒)和一个带负电荷的C端组成,C端是谷氨酸和天冬氨酸的重复链。人类HMGB1是一个包含215个氨基酸的蛋白质,由位于染色体13q12.3的基因编码。HMGB1包含两个DNA结合域:A盒(氨基酸9-79)和B盒(氨基酸95-163),以及一个C末端(氨基酸186-215)。HMGB1含有两个核定位序列(nuclear localization sequence,NLS),分别位于氨基酸28-44(NLS1)和179-185(NLS2)上,负责HMGB1的核定位,并调节HMGB1在细胞核和细胞质之间的转录后修饰,如磷酸化和乙酰化[5]。见图1。
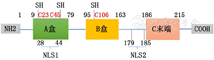
HMGB1在C23、C45和C106氨基酸上有三个关键的半胱氨酸残基,其氧化可以直接影响HMGB1亚细胞定位,这决定了HMGB1是作为细胞因子、趋化因子还是非活性蛋白发挥作用[6]。
HMGB1包含两个NLS(NLS1和NLS2)。HMGB1通过组蛋白乙酰转移酶家族蛋白和组蛋白去乙酰化酶家族蛋白介导的乙酰化和去乙酰化修饰NLS1和NLS2,在细胞核和细胞质之间穿梭[7]。在NLS的翻译后乙酰化或磷酸化,核HMGB1被转移到胞浆中并隔离到细胞质囊泡中。这些细胞质内的HMGB1囊泡可以通过细胞坏死分泌到细胞外空间。除此之外,其他修饰,如甲基化、磷酸化和氧化等也可以调节HMGB1的转运和释放。核HMGB1也可以在Lys42位点甲基化,这可以改变HMGB1蛋白的构象,降低HMGB1-DNA的结合活性,从而增加了其从细胞核向细胞质的被动扩散[11]。胞质HMGB1被蛋白激酶C磷酸化可以阻断其入核,使其留在胞质中[12]。
HMGB1通过其受体触发下游活动。到目前为止,已有十几种受体被认为可以传递HMGB1信号,包括toll样受体(toll-like receptors,TLRs)、晚期糖基化终产物受体(the receptor for advanced glycation end products,RAGE)、syndecan-1(分化簇CD138)、巨噬细胞受体1、趋化因子受体4(C-X-C motif chemokine receptor 4,CXCR4)、触珠蛋白、磷脂蛋白酪氨酸磷酸酶、T细胞免疫球蛋白黏蛋白3、血栓调节蛋白、热稳定抗原/HSA/CD24、骨形态发生蛋白受体2(bone morphogenetic protein-2,BMPR2)和N-甲基-D-天冬氨酸受体等[13,14]。HMGB1除了与这些受体直接相互作用外,还可以与其他免疫共激活因子相互作用形成异质复合物[15]。例如,通过与IL-1β、基质细胞衍生因子1(C-X-C motif chemokine ligand 12,CXCL12)、核小体、DNA、RNA和LPS等结合后,激活其下游信号转导通路诱发各种生理紊乱。
在PH中,肺动脉三层均有变化,包括外膜增厚伴成纤维细胞增殖和免疫细胞募集,中膜增厚主要由平滑肌细胞肥大引起,内膜增厚可归因于内皮下间隙增厚和内皮细胞肥大及增生。Pugliese等[16]表明缺氧和流动/剪切壁应力变化联合引发的炎症级联反应介导了上述效应。而HMGB1就在此过程中扮演了一定的角色。
如前文所述,HMGB1的生物学活性取决于其细胞位置、环境和翻译后修饰。在细胞核内,HMGB1在DNA复制、转录、染色质重构、V(D)J重组等过程中发挥关键作用,从而作为DNA伴侣调控DNA损伤修复和维持基因组稳定性。细胞质HMGB1通过增加自噬、抑制凋亡、参与非常规蛋白的分泌通路[6]和调节线粒体功能[17]参与免疫应答。在细胞膜上,HMGB1促进轴突萌发和轴突生长,激活血小板,诱导细胞迁移。而分泌到胞外的HMGB1具有多种活性,参与炎症、免疫、迁移、侵袭、增殖、分化、抗菌防御和组织再生[6]。
最近研究表明[18,19],PH的发展与肺自噬的增加及抑制凋亡有关,而Lin等[20]的研究发现,用人类同源抵抗素条件培养的人肺微血管内皮细胞可以以HMGB1依赖性的方式诱导人肺动脉平滑肌细胞自噬反应、BMPR2缺陷和随后的抗凋亡增殖,从而协同促进PH的形成。
一些代谢和(或)线粒体过程的中断,如糖酵解开关和氧化磷酸化,以及线粒体生物发生和质量控制的变化,与PH的发展有关[21]。线粒体动力相关蛋白1(dynamin-related protein 1,Drp1)在调节线粒体裂变中发挥重要作用。在人类肺动脉平滑细胞(pulmonary arterial smooth muscle cells,PASMCs)中,Drp1被发现是细胞周期检查点的关键,其过表达被假设是导致细胞过度增殖的原因[22]。在PH大鼠模型中,右心室成纤维细胞Drp1表达增加,抑制其表达可导致右心室成纤维细胞增殖和胶原生成减少[23]。Boytard等[24]表明,TLR4被HMGB1激活可上调RIPK3的磷酸化水平。RIPK3激活磷酸化Drp1,从而触发线粒体裂变,增加线粒体活性氧产生。最近,研究者发现受损细胞释放的HMGB1,通过激活ERK1/2(细胞外信号调节激酶1/2)通路和自噬激活,导致Drp1磷酸化和分裂[25],进而促进PH的发展。
在细胞膜上的HMGB1,可以促进血小板的活化,活化的血小板又可以促进HMGB1释放,形成正反馈循环。活化的血小板可以诱导中性粒细胞胞外陷阱(neutrophil extracellular traps,NETs)的形成,同时血小板的活化及内皮黏附可以促进炎症细胞涌入血管,导致肺血管重构[26]。
而发挥作用的更多的是释放到胞外的HMGB1,发挥其损伤相关模式分子的作用。前文已经提到,有多种受体可以传递HMGB1的信号,但在PH中,谈及最多的是TLRs及RAGE。国内外已经有大量的研究证实了HMGB1与TLRs及RAGE结合,通过一系列的分子机制及信号通路,包括MAPKs、PI3K/Akt,和核转录因子-κB(nuclear factor-κB和NF-κB信号传导通路,促进促炎因子和促血管生成因子的释放,直接或间接的促进肺血管重构及肺动脉高压的形成[27,28]。本文着重提及近几年新发现的一些通路。研究发现,HMGB1通过与TLR4相互作用诱导NETs形成,而HMGB1介导的中性粒细胞清除病原体相关死亡可能是血小板源性HMGB1的促血栓形成作用的机制之一[29]。NETs通过髓过氧化物酶/过氧化氢/NF-κB/TLR4信号通路,在体外和体内触发肺血管内皮细胞的炎症激活,促进血管生成[2]。Xie等[30]发现,肺动脉内皮细胞铁死亡通过炎症小体HMGB1/TLR4/NLRP3信号通路,触发炎症反应从而导致肺血管重构。而Li等[31]发现双链RNA依赖蛋白激酶通过招募与凋亡相关的含CARD的斑点状蛋白与炎症小体结合促进炎症因子的释放。斑点状蛋白作为一个不可或缺的炎症小体接头,在激活包括NLRP3、NLRC4、AIM2和NLRP9b在内的炎症小体中起重要作用,随后炎症小体激活半胱氨酸天冬酶1释放IL-1β和HMGB1,促进PASMCs过度增殖,从而导致PH的发生。同时他们发现蛋白激酶R的缺失可以防止PASMCs在PH发育过程中过度增殖导致的血管重塑。Dai等[32]发现HMGB1通过TLR4和TRPC(瞬时受体电位阳离子通道蛋白)相关的Ca内流以及Akt磷酸化驱动的PASMCs迁移在PH中发挥作用。
除此之外,也有相关研究发现,HMGB1可以抑制BMPR2信号通路促进缺氧诱导的PH。Wang等[33]发现在体外培养的PASMCs中,HMGB1显著促进了PASMCs的增殖和迁移,下调了BMPR2信号通路中p-Smad1/5/8(磷酸化信号转导蛋白1/5/8)和Id1(分化抑制因子1)的表达,其可通过HMGB1抑制剂沙奎那韦、甘草酸和TLR4抑制剂TK-242消除。
TLRs与RAGE并不是单独的发挥作用,RAGE与TLRs在HMGB1诱导的炎症中发挥着协同作用。Zhong等[28]发现,HMGB1可能与细胞表面RAGE受体结合,导致MAPK激活,从而促进TLR4易位到细胞表面,TLR4还可以与HMGB1结合,导致RAGE的转录和翻译,从而促进RAGE易位到细胞表面,与胞外的HMGB1相互作用。RAGE和TLR4相互作用,并随后与HMGB1结合,导致炎症细胞因子的转录和分泌,进而促进PH的发生发展。
尽管在过去二十年中已经探索了许多治疗PH的方法,但目前可用的治疗方法基本上仍然是姑息性的。这就需要进一步的研究来了解肺血管重构的潜在关键调控因子,这可能有助于设计新的PH治疗方法。而近几年HMGB1出现在大家的视野中,将其作为靶点治疗PH已经成了近年来研究的热点。
丙酮酸乙酯可以抑制HMGB1的磷酸化及释放,Liu等[34]通过研究细胞和动物模型表明,丙酮酸乙酯通过抑制HMGB1/RAGE轴进而抑制肺动脉平滑肌细胞的增殖,从而缓解PH。甘草酸可以直接与HMGB1结合,干扰其与受体及免疫共激活因子的结合。相关研究表明,甘草酸阻断HMGB1可减轻大鼠缺氧暴露和野百合碱处理后引起的PH[32]。新型肽P5779特异性靶向细胞外HMGB1,破坏了其与TLR4接头MD2的相互作用,从而抑制HMGB1-TLR4信号通路,为治疗PH提供了一条新的道路与方案[35]。白藜芦醇在缺血缺氧性脑损伤中可以通过激活沉默调节蛋白1降低HMGB1/TLR4/MyD88/NF-κB信号通路和随后的神经炎症反应[36],而白藜芦醇是血管平滑肌细胞增殖的抑制剂,在动物模型中有效延缓PH的进展[37]。乙酰胆碱可以抑制HMGB1的主要受体TLR4和RAGE。Yang等[38]证明,在培养的巨噬细胞中,乙酰胆碱和烟碱乙酰胆碱受体亚型α7激动剂限制了HMGB1和HMGB1-LPS复合物细胞内化。Zi等[39]证明,在右美托咪定治疗实验性脓毒血症中,通过激活α7烟碱乙酰胆碱受体下调TLR4表达,从而减轻炎症反应。而烟碱乙酰胆碱α7受体的活化可以通过抑制HMGB1/TLR4/NLRP3炎性小体减轻野百合碱诱导的大鼠PH[40]。Terameprocol(TMP)是去甲二氢愈创木酚酸的甲基化衍生物;在体内,TMP显著降低了PH和心脏重构的发生,进而改善心脏功能。在体外,TMP抑制PASMCs增殖并诱导其凋亡。Nogueira-Ferreira等[41]研究TMP的抗增殖作用可以通过下调HMGB1来介导。
除了直接抑制HMGB1以及其相关受体,靶向HMGB1与其他免疫共激活因子形成的异质复合物也是一个新的方向。前文提到,HMGB1可与CXCL12形成异质复合物,并通过其受体CXCR4发出信号。研究表明,双氟尼柳在体外和体内靶向HMGB1/CXCL12异质复合物,降低了HMGB1的浓度,并阻断了免疫细胞募集,为HMGB1的新型抑制剂提供了一种新的思路[42]。
PH的发病机制涉及多个方面,炎症机制扮演着重要的角色。HMGB1是一种核蛋白,正常情况下其在胞核中与DNA结合,参与DNA修复和复制。而在应激条件下,其释放到胞质及胞外发挥不同的作用。近年来,有很多的临床前研究证明HMGB1在PH的发生发展中扮演着一定的角色,在实验性PH的治疗中,靶向HMGB1展示了一定的潜能。目前其具体机制尚不明确,需要进一步的研究与探索,从而更好及更安全地运用到临床试验中去,并给诊断与治疗PH提供一条新的思路与途径。
所有作者声明无利益冲突

























