
探讨中国癫痫患者基因突变与临床表现的相关性。
收集2014年1月至2016年7月在首都医科大学宣武医院就诊的特发性癫痫患者23例,发病年龄8个月至31岁。利用二代测序技术,筛查与癫痫相关的基因突变,部分突变通过家系内连续两代患者的Sanger测序得以验证。比较不同基因突变的携带者在癫痫主要临床特点(家族史、性别、发病年龄、发作形式、并发症)方面的差异。
在23例患者中共检出与癫痫发病可能相关的突变基因38个,这些患者多为儿童及青年发病,发病形式以强直阵挛为主,多不伴有智力障碍,病程良性。致病基因以离子通道、酶类、特殊蛋白类基因为主。携带离子通道和特殊功能蛋白基因突变的患者在性别、发病年龄、发作形式、家族史等临床特征方面差异无统计学意义(均P>0.05),但强直-阵挛的患者中检出离子通道基因突变的比例(15/15)高于特殊功能蛋白基因突变携带者(16/20)(P=0.066)。
二代测序技术是检测各类癫痫患者基因突变的有效手段,可用于临床辅助病因诊断。强直-阵挛发作形式可能与离子通道基因突变存在关联性。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
癫痫是神经系统常见的慢性疾病之一,严重影响患者的生活质量。癫痫从病因学可分为遗传性、结构性、代谢性、免疫性、感染性及原因不明。有研究表明,超过50%的癫痫存在遗传基础[1],其中多数为复杂遗传致病,少部分为单基因遗传致病。以往关于癫痫致病基因的研究多数以基因谱系疾病及癫痫综合征致病基因分析为主。本研究采用高通量二代测序新技术,对有癫痫家族史和散发特发性癫痫患者,进行基因突变筛查,为今后癫痫病因学研究和个体化治疗提供帮助。
通过首诊现场和电话回访详细询问、记录病史及家族史等方法,收集2014年1月至2016年7月在首都医科大学宣武医院就诊的中国汉族癫痫患者23例。其中男14例,女9例;就诊年龄6~40岁。17例患者有家族史,6例散发病例。病例纳入标准:符合2005年国际抗癫痫联盟制定的癫痫诊断标准,且遗传因素为主要病因。排除标准:排除症状性癫痫,隐源性癫痫及拒绝基因检测、随访的患者。排除遗传性代谢性疾病合并癫痫的患者。排除已明确癫痫病因的继发性癫痫以及青少年肌阵挛癫痫。排除1例基因检测结果为SCN5A的患者:临床表现为抽搐、猝死,结合突变基因型,最后诊断为长Q-T间期综合征。本次研究患者及其家属均知情同意,且经宣武医院伦理委员会批准。
提取外周血5 ml制备DNA样本-4 ℃保存,采用商业化定制的癫痫及相关发作性疾病基因目标序列捕获测序包(包含CASR、GABRD、CACNA1H、SLC12A5、KCNMA1、SLC2A1、GABRG2、SCN9A、SCN1B等共480个基因)捕获建库,采用X10二代测序仪进行高通量测序。检测到的位点采用polyphen 2积分预测其蛋白功能影响(分为肯定致病、可能致病、不致病或意义不确定等几类)。对疑似突变或部分有连续二代家族成员患病者,采用Sanger测序进行验证。
比较不同基因突变携带者在癫痫主要临床特点(家族史、性别、发病年龄、发作形式、并发症)方面的差异。对频率参数采用χ2检验,连续数值变量采用t检验。P<0.05为差异有统计学意义。所有统计学分析均采用SPSS 17.0软件进行分析。
23例患者均属特发性癫痫。其中17例有明确的家族史,6例为散发病例。男14例,女9例。23例患者中2例有热惊厥史,4例有智力障碍,均无产伤窒息史。首发年龄0~20岁的18例,21~30岁4例,30岁以上1例(31岁),发病年龄集中于青少年。发作形式有失神、强直-阵挛、单纯部分性发作三种。其中失神发作2例,肌阵挛3例、部分性发作4例,强直-阵挛发作19例,所有强直阵挛发作及失神发作均伴有意识丧失。
本组病例共检测出38个突变基因,其中电压门控离子通道11个,配体门控离子通道4个,特殊功能蛋白类基因20个,其他调控因子相关基因3个;基因突变主要分布在离子通道和特殊功能蛋白。本组病例发现同一基因突变患者临床表型可能完全不同,且有13例发现单一基因突变,其余10例均发现2~4个突变基因,部分病人做家系验证后仍考虑存在多个致病基因,下一步需要做全外显子或全基因组检测进一步验证致病基因。这些突变基因既往文献报道与癫痫、热惊厥、共济失调等疾病有关。基因有害性预测Polyphen2>0.85为预测有害变异,本组病例有17例Polyphen2>0.85,突变与癫痫发作明显相关(表1)。
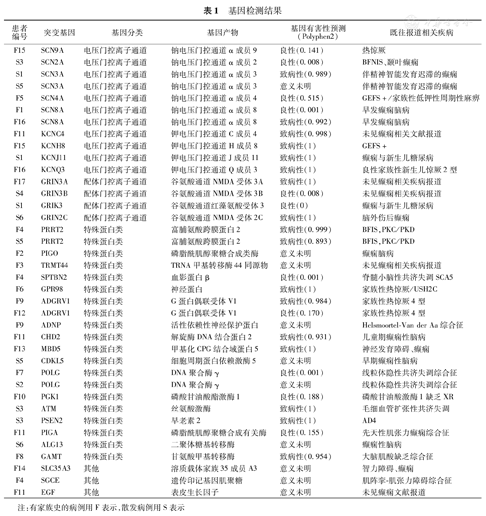
基因检测结果
基因检测结果
| 患者编号 | 突变基因 | 基因分类 | 基因产物 | 基因有害性预测(Polyphen2) | 既往报道相关疾病 |
|---|---|---|---|---|---|
| F15 | SCN9A | 电压门控离子通道 | 钠电压门控通道α成员9 | 良性(0.141) | 热惊厥 |
| S3 | SCN2A | 电压门控离子通道 | 钠电压门控通道α成员2 | 良性(0.008) | BFNIS、颞叶癫痫 |
| S1 | SCN3A | 电压门控离子通道 | 钠电压门控通道α成员3 | 致病性(0.989) | 伴精神智能发育迟滞的癫痫 |
| S5 | SCN3A | 电压门控离子通道 | 钠电压门控通道α成员3 | 意义未明 | 伴精神智能发育迟滞的癫痫 |
| F5 | SCN4A | 电压门控离子通道 | 钠电压门控通道α成员4 | 良性(0.515) | GEFS+/家族性低钾性周期性麻痹 |
| F1 | SCN8A | 电压门控离子通道 | 钠电压门控通道α成员8 | 良性(0.001) | 早发癫痫脑病 |
| F16 | SCN8A | 电压门控离子通道 | 钠电压门控通道α成员8 | 致病性(0.992) | 早发癫痫脑病 |
| F11 | KCNC4 | 电压门控离子通道 | 钾电压门控通道C成员4 | 致病性(0.998) | 未见癫痫相关文献报道 |
| F15 | KCNH8 | 电压门控离子通道 | 钾电压门控通道H成员8 | 致病性(1) | GEFS+ |
| S1 | KCNJ11 | 电压门控离子通道 | 钾电压门控通道J成员11 | 致病性(1) | 癫痫与新生儿糖尿病 |
| F16 | KCNQ3 | 电压门控离子通道 | 钾电压门控通道Q成员3 | 致病性(1) | 良性家族性新生儿惊厥2型 |
| F17 | GRIN3A | 配体门控离子通道 | 谷氨酸通道NMDA受体3A | 致病性(1) | 未见癫痫相关疾病报道 |
| S4 | GRIN3B | 配体门控离子通道 | 谷氨酸通道NMDA受体3B | 良性(0.008) | 未见癫痫相关疾病报道 |
| S1 | GRIK3 | 配体门控离子通道 | 谷氨酸通道红藻氨酸受体3 | 良性(0) | 癫痫与新生儿糖尿病 |
| S6 | GRIN2C | 配体门控离子通道 | 谷氨酸通道NMDA受体2C | 致病性(1) | 脑外伤后癫痫 |
| F4 | PRRT2 | 特殊蛋白类 | 富脯氨酸跨膜蛋白2 | 致病性(0.999) | BFIS,PKC/PKD |
| S5 | PRRT2 | 特殊蛋白类 | 富脯氨酸跨膜蛋白2 | 致病性(0.893) | BFIS,PKC/PKD |
| F2 | PIGO | 特殊蛋白类 | 磷脂酰肌醇聚糖合成类酶 | 意义未明 | 癫痫脑病 |
| F3 | TRMT44 | 特殊蛋白类 | TRNA甲基转移酶44同源物 | 意义未明 | 未见癫痫相关疾病报道 |
| F4 | SPTBN2 | 特殊蛋白类 | 血影蛋白β | 良性(0.001) | 脊髓小脑性共济失调SCA5 |
| F6 | GPR98 | 特殊蛋白类 | 神经蛋白 | 致病性(1) | 家族性热惊厥/USH2C |
| F9 | ADGRV1 | 特殊蛋白类 | G蛋白偶联受体V1 | 致病性(0.984) | 家族性热惊厥4型 |
| F12 | ADGRV1 | 特殊蛋白类 | G蛋白偶联受体V1 | 良性(0.170) | 家族性热惊厥4型 |
| F9 | ADNP | 特殊蛋白类 | 活性依赖性神经保护蛋白 | 意义未明 | Helsmoortel-Van der Aa综合征 |
| F11 | CHD2 | 特殊蛋白类 | 解旋酶DNA结合蛋白2 | 致病性(0.931) | 儿童期癫痫性脑病 |
| F13 | MBD5 | 特殊蛋白类 | 甲基化CPG结合域蛋白5 | 致病性(1) | 神经发育障碍、癫痫 |
| S5 | CDKL5 | 特殊蛋白类 | 细胞周期蛋白依赖激酶5 | 意义未明 | 早期癫痫性脑病 |
| F7 | POLG | 特殊蛋白类 | DNA聚合酶γ | 良性(0.001) | 线粒体隐性共济失调综合征 |
| S2 | POLG | 特殊蛋白类 | DNA聚合酶γ | 意义未明 | 线粒体隐性共济失调综合征 |
| F10 | PGK1 | 特殊蛋白类 | 磷酸甘油酸酯激酶1 | 良性(0.188) | 磷酸甘油酸激酶1缺乏XR |
| S3 | ATM | 特殊蛋白类 | 丝氨酸激酶 | 致病性(1) | 毛细血管扩张性共济失调 |
| S3 | PSEN2 | 特殊蛋白类 | 早老素2 | 致病性(1) | AD4 |
| F11 | PIGA | 特殊蛋白类 | 磷脂酰肌醇聚糖合成有关酶 | 良性(0.155) | 先天性肌张力癫痫综合征 |
| S6 | ALG13 | 特殊蛋白类 | 二聚体糖基转移酶 | 意义未明 | 癫痫性脑病 |
| F8 | GAMT | 特殊蛋白类 | 甘氨酸甲基转移酶 | 致病性(0.954) | 大脑肌酸缺乏综合征 |
| F14 | SLC35A3 | 其他 | 溶质载体家族35成员A3 | 意义未明 | 智力障碍、癫痫 |
| F4 | SGCE | 其他 | 遗传印记基因肌聚糖 | 意义未明 | 肌阵挛-肌张力障碍综合征 |
| F11 | EGF | 其他 | 表皮生长因子 | 意义未明 | 未见癫痫文献报道 |
注:有家族史的病例用F表示,散发病例用S表示
通过比较不同突变携带者的性别、发病年龄、发作形式(是否强直阵挛发作)、是否伴有智力障碍及是否有家族史等比较,均P>0.05,即临床特征与基因型未显现出明显相关性。但从临床资料观察,部分临床表型呈现明显偏态分布,例如:强直-阵挛这一发作形式与基因突变关联性分析P为0.06,提示基因突变可能与强直阵挛发作形式存在关联性(表2)。
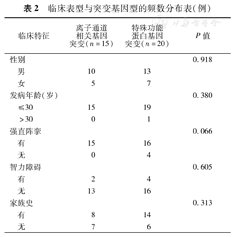
临床表型与突变基因型的频数分布表(例)
临床表型与突变基因型的频数分布表(例)
| 临床特征 | 离子通道相关基因突变(n=15) | 特殊功能蛋白基因突变(n=20) | P值 | |
|---|---|---|---|---|
| 性别 | 0.918 | |||
| 男 | 10 | 13 | ||
| 女 | 5 | 7 | ||
| 发病年龄(岁) | 0.380 | |||
| ≤30 | 15 | 19 | ||
| >30 | 0 | 1 | ||
| 强直阵挛 | 0.066 | |||
| 有 | 15 | 16 | ||
| 无 | 0 | 4 | ||
| 智力障碍 | 0.605 | |||
| 有 | 2 | 4 | ||
| 无 | 13 | 16 | ||
| 家族史 | 0.313 | |||
| 有 | 8 | 14 | ||
| 无 | 7 | 6 | ||
本组病例均符合特发性癫痫特征,发病年龄以儿童及青少年为主,发作形式以强直-阵挛为主,发作多伴有意识不清,多数有家族史,多数病例不伴有智力障碍,病程均呈良性进展。共检测出突变基因38个,突变主要分布在离子通道相关基因和特殊蛋白类基因。本组资料6例散发病例均检出突变基因,提示不伴有家族史的病人仍然有筛查基因突变的必要。收集病例过程中有1例以意识不清、四肢抽搐就诊患者,初诊癫痫,随访过程中再次发作抽搐并猝死,基因检测结果为SCN5A,结合突变基因型及基因功能分析,最后诊断为长Q-T间期综合征,考虑为死亡原因;可见基因检测对修正临床诊断有重要作用。本组病例还发现同一基因突变患者临床表型可能完全不同,且同一患者可能存在多个致病基因,提示环境、成长经历等因素可能和基因突变共同影响临床表型,或者部分特发性癫痫为多基因病,其临床表型受多基因共同作用。
本组病例重复率最高的突变基因为钠离子通道相关基因,共7个;其次是钾离子通道相关基因4个,谷氨酸受体相关基因4个。既往文献显示,目前发现的癫痫相关易感基因有70多种,其中大部分表达的是离子通道或离子通道调节因子[2],本次基因检测结果与国内外报道相符。本组病例中重复率最高的是钠离子通道相关基因:SCN9A、SCN2A、SCN3A、SCN4A、SCN8A。电压依赖型钠离子通道是一类跨膜蛋白,由一个构成中央孔区的α亚基和一个或多个辅助β亚基组成。钠通道α亚单位有不同亚型,包括Nav1.1~Nav1.9,分别由SCN1A, SCN2A, SCN3A, SCN4A, SCN5A,SCN8A, SCN9A, SCN10A, SCN11A基因编码。SCN4A主要在骨骼肌上表达,SCN5A主要在心肌或者神经组织内表达,SCN1A,SCN2A,SCN3A ,SCN8A四个基因主要在脑内表达[3,4,5]。钠通道主导着动作电位的产生和传播,并且影响突触整合和阈下电活动[6],任何微小的钠电流变化都可能对神经元的兴奋性产生重大影响,导致癫痫发作。目前国内外报道与钠离子通道相关的疾病有GEFS+、FS、PEFS+、 SMEI、Lennox-Gastaut综合征等[7,8]。本组病例发现谷氨酸受体相关基因4个,为配体依赖型离子通道基因。本组检测到的GRIN3A、GRIN3B、GRIK3、GRIN2C基因均表达谷氨酸受体蛋白不同的亚基,未见相关的癫痫综合征报道[9,10]。本次研究发现编码酶类及特殊功能蛋白的基因突变20个,证明功能蛋白相关的基因突变在癫痫发生中亦占有重要位置。本研究发现2个PRRT2基因突变,该基因编码富含富脯氨酸的跨膜蛋白,既往报道与PKD、BFIS、ICCA、PNKD、PED、EA相关[11]。
本次研究临床表型与基因型相关性分析中均P>0.05,其中强直-阵挛发作与基因突变关联性分析P值为0.06,显示基因突变与强直-阵挛发作形式存在相关趋势。所以,不同临床表型的癫痫在基因突变筛查中应有所侧重,以强直阵挛为主要发作形式的癫痫应积极筛查致病基因,筛查范围重点为离子通道及特殊蛋白类基因。本次分析显示,任何年龄及性别的癫痫患者均有可能存在基因突变,没有家族史的患者也应该做基因筛查。
总之,本研究表明特发性癫痫有高度遗传异质性,致病基因可能主要改变离子通道和特殊蛋白功能,基因检测在临床诊断中有一定价值。目前的二代测序方法检测到的结果在癫痫病因学的意义,需要更多的样本和更深入的基因功能学研究来证实。随着基因测序成本的下降和平台的成熟,未来癫痫的单基因检测会被更准确、全面的全基因组或全外显子检测替代,也可能研发出新型靶向抗癫痫药物或实现基因编辑等新的癫痫治疗方法。本组资料是我们在近2年临床诊疗过程中随机送检的病例,提供了初步的二代测序技术所能检测到的可能致病基因,但是本组病例数量过小,今后研究应进一步扩大随机样本量。

























