
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
例1 女,3岁5月龄,汉族。因"反复皮肤瘀斑伴鼻衄3年余"于2016年3月在贵州省儿童医院就诊。患儿母亲为孕2产2,患儿生后1个月发病,6月龄后明显,外伤或剧烈活动后皮肤瘀斑加重,时有鼻衄。多次就诊于省内外多家医院检查有贫血,血小板数量正常,凝血功能正常,诊断不明,曾4次输红细胞治疗。父母为姑表亲婚配,哥哥有类似病史,5岁时便血而死亡。轻度贫血貌,全身皮肤见散在出血点和瘀斑,肝脾淋巴结无肿大,心肺腹及神经系统查体无异常。入院后辅助检查:白细胞(WBC)10.63×109/L,中性粒细胞(N)0.43,淋巴细胞(L)0.47,红细胞(RBC)2.36×1012/L,血红蛋白(HB)67g/L,红细胞比容(HCT)0.20,血小板(PLT)127×109/L,网织红细胞百分比3.6%,网织红细胞绝对值73.0×109/L。血涂片血小板分散,不聚集。电解质、肝肾功能未见异常。出血时间延长25 min,血块收缩不良,凝血功能正常;流式细胞仪检测血小板CD41阳性率0.7%,CD61阳性率1.3%;血液遗传病基因筛查(第2代高通量测序,送北京康旭医学检验所检测),患儿17号染色体ITGA2B基因4号外显子c.480C>G:p.S160R纯合突变;父母均有该位点的杂合突变。诊断遗传性血小板无力症,输注红细胞纠正贫血,输注血小板后鼻出血停止,住院4 d好转出院,出院后遵医嘱避免外伤。门诊随访1年皮肤和鼻出血症状减轻,未再发生失血性贫血。
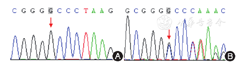
例2 女,12岁,汉族。因"月经初潮后出血不止10 d"于2016年7月在贵州省儿童医院就诊。患儿母亲为孕2产2,4岁后即出现鼻衄,感冒、外伤或剧烈活动后出现皮肤瘀斑和鼻衄,多次就诊于当地医院,诊断不明。父母姑表亲婚配,哥哥有类似的病史(未就诊)。体查:轻度贫血貌,全身皮肤见散在出血点和瘀斑,肝脾淋巴结无肿大,心肺腹及神经系统查体无异常。入院后辅助检查:WBC 12.21×109/L,N 0.72,L 0.22, RBC 2.5×1012/L,HB 61 g/L,HCT 0.20,PLT 251×109/L,网织红细胞未查,血涂片血小板分散,不聚集。电解质、肝肾功能未见异常。出血时间延长18 min ,血块收缩不良,凝血功能正常;流式细胞仪检测CD41阳性率31.3%,CD61阳性率41.0%;患儿和父亲(父母离异,母亲未能联系到)的血液遗传病基因筛查(第2代高通量测序,送海斯特医学检验所检测),患儿17号染色体ITGA2B基因20号外显子c.2077dupG:p.A693F纯合突变(图1A),父亲为杂合突变(图1B)。诊断遗传性血小板无力症,输注红细胞纠正贫血,输注血小板止血,妇科会诊使用缩宫素和去氧孕烯炔雌醇片,5 d后阴道出血停止出院。门诊随访半年,月经期未再出现严重出血。
遗传性血小板无力症是1种少见的常染色体隐性遗传性出血性疾病,由于血小板膜糖蛋白Ⅱb/Ⅲa基因缺陷所致,GPⅡb(αⅡb,CD41)和GPⅢa(β3,CD61)分别由ITGA2B和ITGB3基因编码,目前已登记的GPⅡb基因突变已经超过190种,GPⅢa基因突变超过120种[1]。依据血小板GPⅡb/Ⅲa含量GT分3型:Ⅰ型GPⅡb/Ⅲa数量少于正常5%;Ⅱ型GPⅡb/Ⅲa数量少于正常25%;Ⅲ型为变异型,GPⅡb/Ⅲa数量几乎正常但质量低下[2]。本例1为Ⅰ型GT,出血程度重,例2为Ⅲ型GT,出血程度轻。2例均以反复皮肤黏膜出血为主要症状,例1鼻衄致失血性贫血,例2月经量多导致失血性贫血,均因中度贫血而入院,说明合并明显贫血可引起家长重视而就诊。但遗传性血小板无力症的出血表现有异质性,临床分型与基因型并不一致,出血的频率和严重程度不一,且与GPⅡb/Ⅲa的含量无关联,患儿可测不到GPⅡb/Ⅲa,只有轻微出血,也可GPⅡb/Ⅲa含量正常但有明显出血。GT患儿通常在7岁前发病,多数为皮肤或黏膜出血、外伤后出血加重,部分患儿表现为胃肠道出血、血尿,少见颅内出血和关节血肿[3]。2例患儿在诊断后,及时输注血小板止血,指导避免剧烈活动和外伤,例2患儿月经期妇科干预,均未再出现严重出血,故对该病只要早期及时诊断、治疗和预防,预后良好。
2例患儿均有多次在多家医院就诊的经历,分别在发病3年余和8年余才明确诊断,说明该病早期诊断困难,与该病较少见,认识不足且缺乏实验室诊断条件有关。结合2例患儿诊治经验,阳性家族史、父母近亲婚配史可提供较强的诊断线索,对自幼起病,且排除血小板减少、血友病等常见的出血性疾病,结合外周血涂片血小板不聚集,首先应考虑该病,进一步做基因检查协助诊断。随着分子生物学进展和基因测序的广泛开展,对该病的完整诊断应该包括基因突变类型,不仅能帮助临床诊断分型,还能指导优生优育。例1患儿因ITGA2B基因4号外显子c.480C>G,发生纯合错义突变,第160位丝氨酸变为精氨酸而致病;例2因20号外显子c.2077dupG发生纯合移码突变,第693位丙氨酸变为苯丙氨酸而致病。国内外报道未检测到本组患儿两种突变[4,5,6],但该突变是否有地区特异性,其对GPⅡb/Ⅲa复合物的合成、转运、表达和功能的影响,有待深入研究。

























