
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿男,系第2胎第2产,胎龄36周剖宫产娩出,出生体重4 200 g,羊水清,6 000 ml,生后即出现发绀,1、5 min Apgar评分为8分、9分,予气管插管呼吸机辅助通气、抗感染、补液等,血氧饱和度0.80~0.90,转运至我院。其母孕期定期产检,孕30周发现羊水过多。患儿胞姐12岁,体健。否认家族遗传病史。入院查体:T 35.8 ℃,P 142次/min,R 62次/min,BP 65/40 mmHg;神清,反应差;面容特殊,额窄口大;右肺呼吸音稍低,心脏无明显异常,肝右肋下4 cm,剑突下3.5 cm,边缘钝,质中。入院后查血常规、甲状腺功能正常。血生化ALT 138 U/L,AST 613 U/L,GGT 606 U/L,TBil 46.9 μmol/L,DBil 8.4 μmol/L,TBA 7.2 μmol/L。胸部X线、CT示先天性右侧膈疝;心脏彩超示室间隔缺损、动脉导管未闭、卵圆孔未闭、肺动脉高压、主动脉瓣返流;颅脑彩超示双侧脑室轻度增宽,脑实质回声片状增强。入院后予以呼吸机辅助呼吸,全麻下行右膈疝修补术,术中发现肝脏巨大,大部分疝入右侧胸腔。术后对症支持治疗1个月后逐渐离氧,逐步增加奶量至135 ml/kg出院。全基因组检测到1个罕见可能致病拷贝数变异,位于Xq26,597Kb,共4个基因,包括GPC3基因7-8外显子。在核心家系内行多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)验证,患儿该片段纯合缺失,母亲杂合缺失,父亲正常。随访患儿出院后吸奶慢,3月龄体重4 kg,4月龄4.5 kg,5月龄5.5 kg。6月龄能抬头、不会翻身,智能发育筛查发育商50分,智商50分。仍可见患儿腭窄且腭弓高、舌中央有纵向切迹及纵沟(图1A),手大呈方形、拇指(趾)增宽、食指指甲发育不良( 图1B)。血甲胎蛋白正常,腹部CT未见肿瘤性病变。
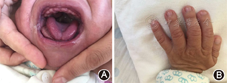
Simpson-Golabi-Behmel综合征(Simpson-Golabi-Behmel syndrome,SGBS)是X连锁隐性遗传病(OMIM:#312870),由于编码磷酸脂酰肌醇聚糖(glypica-3)的基因GPC3发生突变,对胚体中胚层组织生长失控而发病,以过度生长并有多发畸形为主要临床表现,1988年被命名[1],患病率至今未知。SGBS常有颅面异常,表现为巨头畸形或颅缝早闭,宽鼻、巨舌、腭窄且腭弓高,唇裂和腭裂,舌中央有纵向切迹或纵沟等[2,3],可出现各种心血管畸形并与SGBS早期的高病死率有关,肝、肾、脾等内脏增大,不足10%的患儿有先天性膈疝,部分患儿因咬合关节畸形、共鸣出现问题造成典型面型及说话困难[4]。患儿手大且呈方形,可有拇指(趾)增宽、第2、3指(趾)软组织并连等[5]。患儿易患Wilms瘤、肝母细胞瘤、神经母细胞瘤、性腺母细胞瘤及肝癌等。
本例患儿为巨大儿、特殊面容、指趾畸形,合并先天性心脏病、右侧膈疝、肝肿大,中国计量认证检测提示Xq26包含GPC3基因1-7外显子区域共597 Kb缺失,MLPA检测证实该缺失,且核心家系内验证发现符合X连锁隐性遗传模式,明确诊断为SGBS。SGBS患儿主要针对新生儿低血糖和各系统畸形进行对症治疗,以及进行各种康复支持,长期随访目的是为早期发现肿瘤并干预。由于临床表现不同,SGBS患儿预后和寿命不同。临床医生需对SGBS加强认识,可开展基因诊断和产前诊断,进一步积累本病基因型与表型相关资料,对提供遗传咨询具有积极意义。

























