
分析白细胞介素10(IL-10)/转化生长因子-β(TGF-β)诱导的巨噬细胞对肾缺血再灌注损伤的影响。
IL-10/TGF-β联合诱导骨髓源性巨噬细胞,获得M2c型巨噬细胞,尾静脉输注给肾缺血再灌注损伤(IRI)模型小鼠。肾IRI后3 d检测肾功能,HE染色分析肾组织病理变化,脱氧核糖核苷酸末端转移酶介导的缺口末端标记法检测肾小管细胞的凋亡,增殖细胞核抗原的免疫荧光染色分析小管细胞增值,聚合酶链反应及流式细胞仪分别分析肾组织炎症因子和调整性T淋巴细胞(Treg)表达,评估M2c巨噬细胞对肾缺血再灌注损伤的影响。
成功获得成熟巨噬细胞(纯度>95%,增殖能力<1%)及M2c型巨噬细胞,与经典活化的M1型比较,M2c表达低水平的MHCⅡ(P<0.01)、CD86(P<0.01)及炎症因子TNF-α(P<0.01)、IL-1β(P<0.01),但却表达高水平的IL-10(P<0.01)。M2c型巨噬细胞可以明显改善肾IRI小鼠肾功能(P<0.01或P<0.05)及病理损伤(P<0.05);并能抑制小管上皮细胞的凋亡(P<0.01),促进肾小管上皮细胞的增殖(P<0.05),减轻肾组织内炎症因子TNF-α(P<0.05)、IL-1β(P<0.01)和IL-6(P<0.05)浸润,增加调节性T淋巴细胞表达(P<0.01)。
IL-10/TGF-β诱导的M2c型巨噬细胞通过抑制小管细胞凋亡、炎症细胞浸润及促进小管细胞增殖等对肾缺血再灌注损伤发挥保护作用。M2c巨噬细胞可能成为干预肾移植等过程导致的肾IRI的新策略。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
缺血再灌注损伤(ischemia reperfusion injury,IRI)是器官移植及多种疾病伴发的必然过程,能导致实质细胞的凋亡、坏死、炎症细胞浸润及纤维化的形成,是器官损伤的主要原因,具有较高的发病率和病死率[1]。巨噬细胞是天然免疫反应细胞,具有吞噬、抗原提呈、分泌一系列细胞因子等功能,连接天然免疫和适应性免疫。其全身分布广泛,如脑小星型胶质细胞、肺巨噬细胞、肝的Kupffer细胞,肾脏巨噬细胞等。当前,多数研究提示,巨噬细胞因环境因素变化而表现出不同的表型及功能,具有明显的异质性[2,3]。经典途径活化的巨噬细胞(classically activated macrophage,M1)和替代途径活化的巨噬细胞(alternatively activated macrophage,M2)是根据TH1和TH2细胞因子体外刺激而产生的促进炎症反应和调整炎症反应、组织重塑等不同功能的巨噬细胞[4,5,6]。白细胞介素10(IL-10)/转化生长因子-β(TGF-β)体外可诱导巨噬细胞向M2c型转化。这种M2c型巨噬细胞具有更强的免疫抑制能力,如抑制T淋巴细胞增殖和炎症因子分泌、诱导调节性T淋巴细胞(Treg)等,体内研究提示此功能M2c可以明显保护阿霉素肾病小鼠肾功能[7,8]。在肾脏的缺血再灌注损伤方面虽有提示,不同表型的巨噬细胞导致了肾脏的损伤(M1型)和修复(M2型)[9],但是针对M2型巨噬细胞输注是否可以保护肾脏缺血再灌注损伤尚不清楚。本研究旨在探讨M2c型巨噬细胞对肾缺血再灌注损伤的影响,现报告如下。
8~10周龄、20~27 g的雄性C57BL/6小鼠,购于西安交通大学实验动物中心,并饲养在无特定病原体级动物房,自由接触水和饲料。
胎牛血清(Gibco公司);DMEM/F12培养基(Hyclone公司);巨噬细胞集落刺激因子(M-CSF,peprotech公司);IL-10、TGF-β(peprotech公司)苏木素伊红(HE)染色试剂盒(博士德公司);羊抗鼠PCNA一抗(abcam公司);Treg检测试剂盒(ebioscience公司);流式抗体:藻红蛋白标记抗鼠F4/80、多甲藻叶绿素蛋白标记抗鼠CD11b、别藻蓝蛋白标记抗鼠主要组织相容性复合物(MHC)Ⅱ、CD86(Biolegend公司);细胞凋亡检测试剂盒(Roche公司);总RNA提取试剂盒(Omega公司);逆转录试剂盒、荧光定量聚合酶链反应(PCR)试剂盒(ABI公司);胶原酶Ⅳ、DNA酶Ⅰ(Worthington公司);小鼠淋巴细胞分离液(Cedarlane公司)。
无菌取C57BL/6小鼠股骨、胫骨,磷酸盐缓冲液(PBS)冲出骨髓,200目滤网过滤,裂解红细胞。获得的骨髓细胞采用DMEM/F12培养基+M-CSF(20 ng/ml)+体积分数10%胎牛血清(FBS)培养,每2~3 d换液1次,去掉悬浮细胞,12 d后得成熟骨髓源性巨噬细胞。其后继续培养2 d为未活化巨噬细胞(M0),加脂多糖(LPS,100 ng/ml)+γ干扰素(IFN-γ,100 U/ml)培养2 d为M1型巨噬细胞,加IL-10(10 ng/ml)+TGF-β(10 ng/ml)培养2 d为M2c型巨噬细胞。
黏附的骨髓细胞培养至第7天时获得的巨噬细胞(常规的培养方法[10])和培养至12 d时获得巨噬细胞,分别CFSE(羟基荧光素二醋酸盐琥珀酰亚胺脂)染色,并继续培养2 d,流式细胞仪分析其增殖情况。采用流式细胞仪按说明书步骤进行不同活化巨噬细胞的表型及Treg测定。
体积分数2%水合氯醛腹腔注射麻醉,0.02 ml/g,肋下脊柱两侧备皮1.5 cm2,切口采用脊柱旁0.5 cm、肋下0.5 cm、平行脊柱的双侧切口,长1 cm。先行左侧切口,提取肾脏,钝性分离肾蒂,暴露血管,无损伤血管夹夹闭左侧肾动脉30 min,使肾脏缺血。同法暴露右肾及血管,0号线结扎血管,切除右肾,间断缝合皮下及皮肤。左肾血管夹闭到规定时间后,去掉血管夹,使肾脏再灌注,见肾脏变红后回纳腹腔,缝合(IRI组)。假手术组(Sham组)除夹闭血管步骤外执行相同的手术过程。IRI+M2c组每只小鼠在IRI后6 h通过尾静脉输注2×106巨噬细胞,IRI组输注等量PBS。
每组5~7只小鼠,肾IRI后3 d处死小鼠,取抗凝全血,室温下400×g离心10 min,取上层血清,全自动生化分析仪测定肾功能指标:血肌酐、尿素氮。取1/3肾组织,于体积分数4%多聚甲醛溶液中固定后石蜡包埋。制成4 μm切片,用于HE染色和凋亡细胞(Tunel染色)及增殖细胞核抗原(PCNA)的免疫荧光染色;其余肾组织用于提取总RNA和检测Treg表达。
每只小鼠4张切片,每张切片随机于400倍视野下选择10个非重复视野。HE染色结果的病理学评分:根据皮髓交界以及外髓质部位的损伤肾小管细胞坏死、小管腔膨胀、微绒毛脱落及管型形成等病理改变的比例进行小管损伤程度评分:0分,未见;1分,损伤程度<10%;2分,10%≤损伤程度<25%;3分,25%≤损伤程度<50%;4分,50%≤损伤程度<75%;5分,75%≤损伤程度≤100%。免疫荧光染色:计数每个400倍视野下阳性的细胞数。
采用E.Z.N.A.®总RNA提取试剂盒(E.Z.N.A.® Total RNA Kit),按其步骤提取不同极化状态的巨噬细胞(M0、M1、M2c)及肾组织总RNA。应用High Capacity模板DNA逆转录试剂盒(High Capacity cDNA Reverse Transcription Kit)进行RNA的逆转录反应获得模板DNA。SYBR Green PCR Master Mix试剂盒进行PCR反应,引物序列见表1。每份样本设3个复孔,采用2-△△T法计算相关基因的相对表达量(肌动蛋白β为内参照)。

实时荧光定量聚合酶链反应的引物序列
实时荧光定量聚合酶链反应的引物序列
| 基因 | 序列 |
|---|---|
| TNF-α | 正向引物5’-GCGGACTCCGCAAAGTCTAAG-3’ |
| 反向引物5’-CGGACTCCGCAAAGTCTAAG-3’ | |
| IL-1β | 正向引物5’-TGCCACCTTTTGACAGTGATG-3’ |
| 反向引物5’-ATGTGCTGCTGCGAGATTTG-3’ | |
| IL-10 | 正向引物5’-CCAGTACAGCCGGGAGACA-3’ |
| 反向引物5’-CAGCTGGTCCTTTGTTTGAAAG-3’ | |
| IL-6 | 正向引物5’-CACAAGTCCGGAGAGGAGAC-3’ |
| 反向引物5’-TTGCCATTGCACAACTCTTT-3’ | |
| β-actin | 正向引物5’-TGAGAGGGAAATCGTGCGTG-3’ |
| 反向引物5’-TTGCTGATCCACATCTGCTGG-3’ |
数据以均数±标准差( ±s)表示,采用IBM SPSS(19.0版)件分析。统计学方法采用t检验(两个独立样本的均数比较)或方差分析(多个样本均数比较)。P<0.05为差异有统计学意义。
±s)表示,采用IBM SPSS(19.0版)件分析。统计学方法采用t检验(两个独立样本的均数比较)或方差分析(多个样本均数比较)。P<0.05为差异有统计学意义。
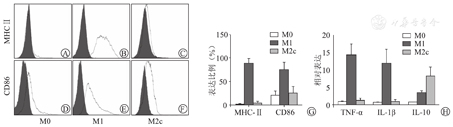
为获得更好同源性的巨噬细胞,选择骨髓源性巨噬细胞。采用Weischenfeldt等方法体外M-CSF刺激培养黏附的骨髓细胞7 d可获得巨噬细胞[10],纯度为97%(图1A)。体外刺激时间可能影响骨髓源性巨噬细胞的成熟性及进一步的功能稳定性。为了验证巨噬细胞的成熟与否,分析其增殖变化,在培养7 d时,巨噬细胞仍然具有较明显的增殖能力(图1B),但培养至12 d时未发现明显增殖(图1C),表明其已成熟,可用于进一步研究。因此,接下来应用IL-10、TGF-β体外刺激48 h,与LPS+IFN-γ刺激的M1型巨噬细胞比较,MHC-Ⅱ、CD86表达明显偏低(P<0.01,图2),炎症因子TNF-α、IL-1β也明显偏低,但IL-10表达明显增加(P<0.01,图2),说明成功获得M2c型巨噬细胞。

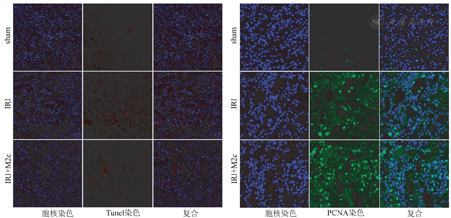
为明确M2c型巨噬细胞是否对肾脏IRI具有保护作用,应用M2c型巨噬细胞输注干预C57BL/6小鼠的肾IRI模型(M2c组),缺血后3 d检测血肌酐(图3A)和尿素氮(图3B),与假手术组比较,模型组(IRI组)肾功能明显异常(P<0.01),而M2c组较IRI组明显降低(P<0.05或P<0.01)。为进一步验证M2c的保护作用,分析组织病理学变化,IRI损伤能引起严重肾小管上皮细胞的凋亡、坏死、扩张及刷状缘脱落(图3D、图3E)等病理变化,但M2c输注明显减轻了这种病理变化(P<0.05,图3F)。因此,我们认为M2c型巨噬细胞能够减轻肾脏缺血再灌注损伤。

肾小管细胞的凋亡、坏死是肾IRI的重要特征,也是导致炎症反应及肾功能异常的主要原因。细胞凋亡、坏死导致染色体DNA断裂,产生3’-OH末端,应用脱氧核糖核苷酸末端转移酶介导的缺口末端标记法(Tunel)标记3’-OH端。发现应用M2c输注后肾小管上皮细胞Tunel染色较模型组明显减少(P<0.01,图4A、图5)。对增殖的影响,应用在细胞增殖的启动上起重要作用的PCNA为标记,反映肾小管细胞的增殖状态。发现M2c在降低小管细胞凋亡的同时,还促进了其增殖,因为较模型组PCNA表达明显增加(P<0.05,图4B、图5)。

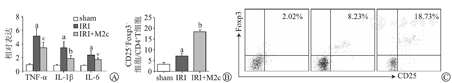
肾IRI后,活性氧及坏死细胞释放的某些蛋白成分等导致炎症系统激活,渗入IRI肾脏,致使肾损伤加重。为进一步明确M2c对IRI的保护作用,观察其对炎症反应的影响。从图3D中,除观察到肾小管细胞的变化外,也能注意到M2c组较IRI组整个炎症细胞浸润明显减少,表明其可以抑制炎症细胞浸润。为进一步证明其对炎症反应的作用,分析肾脏内炎症因子的表达变化。qPCR分析炎症因子TNF-α、IL-1β、IL-6 mRNA变化,发现M2c组三者表达均明显降低(P<0.05或P<0.01,图6A)。为了探讨在IRI环境下,M2c对Treg的影响,检测了IRI肾组织内Treg表达,发现在IRI后3 d Treg表达增加(P<0.01,图6B、图6C),这可能与IRI引起的炎症反应抑制及后期组织修复有关。
巨噬细胞全身分布广泛,但不同组织、器官的巨噬细胞具有不同的性质,同一组织或器官不同时期 的巨噬细胞也可能具有明显的表型及功能等不同,同一组织器官同一时期的巨噬细胞也可能具有明显不同的性质功能[2]。这既是巨噬细胞多功能的体现,同时也为巨噬细胞研究带来困难。为了能更好的研究M2c型巨噬细胞对IRI的作用,我们选用具有更好同一性的骨髓源性巨噬细胞作为细胞来源[10]。骨髓源性巨噬细胞体外通过刺激因子促进前体细胞分化为巨噬细胞。常规方法是巨噬细胞集落刺激因子体外刺激骨髓细胞7 d获得巨噬细胞,进行下一步研究探索[10,11]。我们在研究中发现,培养7 d的巨噬细胞仍然具有一定的增殖特性,而成熟的巨噬细胞为终末分化的细胞,不再具有增殖潜能,所以培养7 d的巨噬细胞仍未完全成熟,这可能影响其功能发挥,因为体外刺激获得M2c后,因其未成熟,输入体内后可能易于失去其抑制及组织损伤修复功能[12]。为此,我们通过延长体外培养时间至12 d,未再发现明显的增殖活动。这也为我们下一步实验奠定了坚实的基础。
巨噬细胞属于天然免疫细胞,通过其分泌细胞因子及抗原提呈等功能连接天然免疫和适应性免疫,在病原微生物入侵过程中加速微生物及受损组织的吞噬、清除,是人体的重要防线。但防御过度或针对正常组织的反应则可导致组织损伤,众多研究提示,炎症反应过程中删除巨噬细胞可以减轻炎症反应,避免组织损伤,在肾脏的IRI过程亦是如此[9,13,14]。但同时也发现,在一定情况下删除巨噬细胞反而会导致组织损伤加重,表明了巨噬细胞的炎症调整及组织修复功能[15,16,17]。在肾脏损伤中巨噬细胞也可能具有上述功能,如在IRI的后期Lee等[9]也观察到巨噬细胞的组织修复功能[18]。但是针对IRI早期的炎症反应及组织损伤,巨噬细胞功能如何呢,促进巨噬细胞向抑制炎症反应及组织修复的M2型转移是否能成为干预IRI的防御措施呢?为此,我们采用输注免疫抑制功能的M2c给小鼠肾IRI模型进行探讨,试验中观察到小鼠肾功能较模型组明显改善,同时组织学检查也发现M2c改善了IRI小鼠肾小管上皮细胞的完整性,进一步确定了其保护作用。巨噬细胞在IRI后即明显浸入受损组织,分泌炎症因子、趋化淋巴细胞渗入,致使炎症反应加重[13,19]。因此,结合我们的研究,进一步探讨如何在IRI过程调整其激活状态、促进其向M2型转变,发挥自身的抗炎、促修复功能,将为IRI治疗提供新方向。
良好的血液供应是保持组织器官正常代谢、维持正常功能的必要条件。多种原因导致的局部缺血,可使细胞、组织及器官发生缺血性损伤(代谢障碍、结构破坏、细胞坏死等)。肾IRI的主要特征之一是因为氧化应激、内质网功能障碍、钙超载等导致的肾小管上皮细胞的凋亡、坏死,坏死的细胞释放正常情况下与免疫系统隔离的各种蛋白成分,如非甲基化的DNA、IL-1α、热休克蛋白、S100蛋白、三磷酸腺苷等,这被称为促炎症损伤相关的分子模式(DAMP),引发炎症反应[20]。因此,缺血、缺氧导致的细胞损伤、坏死不但是结果,更是导致损伤加重的因素。因此,为探讨M2型巨噬细胞在肾IRI中减少组织损伤的机制,我们首先探讨了其对肾小管上皮细胞的凋亡与增殖影响。我们观察到,应用M2c的IRI模型的肾小管上皮细胞凋亡明显减少,而且其增殖也相应增加,这应该是M2c保护IRI的机制之一。在其他组织中,M2型巨噬细胞抑制细胞凋亡、促进细胞增殖也有报道[21,22]。在肾脏IRI损伤后期,肾小管上皮细胞分泌的相关因子可促进巨噬细胞向M2型转变,进而再促进自身的增殖修复[9]。上述报道与我们的研究相一致,进一步证实了M2型巨噬细胞可以抑制小管细胞凋亡、促进增殖的作用。而M2型巨噬细胞抑制肾小管上皮细胞凋亡、促进增殖的机制,可能与其分泌的IL-10相关,因为应用腺病毒为载体高表达IL-10的巨噬细胞可以促进肾小管细胞的增殖[23]。但M2c型巨噬细胞除分泌高水平的IL-10外,还表达其他可能参与抑制细胞凋亡、促进增殖的相关因子,如精氨酸酶Ⅰ等[24]。
炎症反应是IRI后的重要表现,同时也是导致组织损伤的重要原因。既有天然免疫反应的参与,又有适应性免疫细胞的渗入,两方面相互作用,炎症反应的逐级放大,导致组织损伤。受损组织及活性氧等募集中性粒细胞、巨噬细胞等炎症细胞浸入,同时分泌TNFα、IL-1β、IL-6等炎症因子,而炎症因子可进一步募集白细胞渗入,加之补体系统、血小板活化等影响,导致组织损伤不断加重,因此抑制炎症细胞聚集及细胞因子释放对减轻IRI损伤具有重要意义。巨噬细胞为天然免疫反应的重要成员,是天然免疫与适应性免疫的桥梁。M2型巨噬细胞在多种模型中均发现免疫抑制功能,抑制炎症反应。而M2c型巨噬细胞具有更明显的免疫抑制功能,可以抑制巨噬细胞的M1型活化、T淋巴细胞的增殖及诱导Treg[7]。但在IRI损伤的早期炎症反应中,M2c功能如何?我们的研究提示,M2c同样可以抑制炎症细胞浸润、炎症因子表达,发挥保护IRI肾脏的作用。T淋巴细胞,特别是CD4+T淋巴细胞在IRI过程中的重要性越来越引起重视。通过应用阻止T淋巴细胞激活、迁移、增殖及免疫缺陷、特异性T淋巴细胞亚集敲除、过继性转移T淋巴细胞等方法已证明辅助性的CD4+T淋巴细胞参与IRI[25,26]。我们在发现M2c减少T淋巴细胞渗入的同时,也观察到Treg增加。这可能与其促进渗入受损肾脏的CD4+T淋巴细胞向Treg转化或促进外周Treg迁移至肾脏有关,还需进一步研究探讨。Treg参与IRI过程已有较多报道,与其他种类炎症反应中其抑制免疫反应的功能相同,他们在IRI中也表现为参与抑制炎症反应,应用CD25单克隆抗体可以明显增加损伤组织炎症细胞浸润,而过继转移Treg则可以减轻炎症反应,保护IRI组织,另外,Treg还可促进损伤组织的修复[27]。所以,M2c对IRI肾脏的保护应该也与其扩增Treg有关。
虽然我们研究没有进一步评价M2c在体内分布、存活时间、表型稳定等问题,但是我们证明了其对肾IRI损伤的保护作用,表明免疫调整性的巨噬细胞不但在肾IRI后期的损伤修复中发挥重要作用,在早期的炎症反应抑制、促进组织修复方面仍然具有重要作用,所以在肾IRI过程中开发针对调整巨噬细胞表型的药物或方法,可能成为干预肾IRI的新方法,但需要针对表型转化机制的深入探索。

























