22q11.2缺失综合征是最常见的染色体缺失综合征。大部分DiGeorge异常患者具有22q11.2的杂合缺失。异常的腭弓发育可导致甲状旁腺、胸腺和心脏圆锥动脉干的发育缺陷。胸腺发育缺陷导致免疫缺陷病。完全性DiGeorge综合征在所有患者中的比例<0.5%,表现为胸腺缺如和严重T淋巴细胞缺陷。大部分患者为部分型,具有可变的T淋巴细胞缺陷。该病表型谱广泛,包括语言发育延迟、神经精神异常和眼、耳、鼻、咽喉、口腔科异常。此类患者自身免疫性疾病风险增加,重症患儿需要胸腺移植。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Lobdell[1]于1959年首次在病理解剖中注意到甲状旁腺和胸腺同时缺失。DiGeorge[2]于1965年开始描述婴儿出现甲状旁腺功能减低、胸腺发育不良和细胞免疫缺陷的组合,被定义DiGeorge综合征。很快,该综合征又扩展为包括特殊面容,先天性心脏病尤其圆锥动脉干异常。高分辨率的细胞遗传学发现90%的患者具有22q11区域1.5~3.0 Mb的杂合缺失[3]。该缺失还与其他表型有关,如腭-心-面综合征、圆锥动脉干面综合征、降口角肌发育不良并室间隔缺损(Cayler syndrome)和先天中线发育异常综合征(Opitz-G/BBB),提示这些异常有共同起源。该区域内的TBX1基因单倍型功能不全也可引起DiGeorge异常(DGA)。DGA还与10p13缺失综合征、眼组织缺损、心脏异常、鼻后孔闭锁、智力迟缓、生殖、耳异常(CHARGE综合征)及糖尿病目前所产婴儿有关。胎儿酒精综合征、孕母维生素A暴露也可引起DGA表型。
目前研究认为发病机制是第三、四咽囊发育缺陷,头神经嵴细胞不能正常移行至此所致。
遗传方式大部分为散发的,来源于新发突变。6%~28%呈常染色体显性遗传,56%为母亲来源,44%为父亲来源。母亲平均怀孕年龄为29.5岁,与健康人群近似。女性22q11.2区域重组率为男性的1.6~1.7倍,该区域减数分裂时重组率增加可能是母源比例高的原因[4]。
最初细胞遗传学研究提示DGA患者存在不同染色体间的移位,或者单体型不平衡移位22pter-q11,或者染色体内缺失del(22)(q11.21-q11.23),因此,推测DGA的关键区域位于22q11。随着分子生物学的进步,针对细胞核型正常的DGA患者进行分子载量分析和荧光原位杂交,发现大部分患者具有22q11区域的杂合缺失[5]。其他缺失还涉及10p13,18q21.33。也有嵌合的22号染色体单体型报道,患者具有DGA的面容特征、肌张力高、关节伸展受限、所有手指呈弯曲收缩状态。
物理图谱显示缺失位于断裂点关键区域的近端。在经典缺失区域鉴定了4个低拷贝重复(LCRs),具有97%~98%的相似性,直接参与22q11缺失的形成。非人灵长类荧光原位杂交(FISH)分析提示重复事件产生LCRs集聚在2.0亿~2.5亿年前已经出现。
DGS患者边缘区域的单体型重建发现近端染色体间的交换明显增加,另一条正常22号染色体间交换出现率为2/15,与遗传距离一致。用MLHI抗体免疫染色,人类精子减数分裂中75%交换定位于22q的远端,也反映遗传距离。与William综合征不同,FISH分析未发现LCRs附近的染色体逆转。减数分裂 Ⅰ 期异常的染色体间交换事件在22号染色体近端区域可能是缺失的原因,小的缺失更常见于家族遗传。
与22q11缺失相关的症状包括180余种。单卵双生子研究表明存在个体间和家族内的变异。缺失的大小与临床表型缺乏相关性。DGA最初的三联征包括先天性无胸腺、甲状旁腺缺如及心脏异常。基于此,临床诊断标准为符合下述4项中的3项:先天性心脏病、特征面容、甲状旁腺功能低下或新生儿低钙、缺失或异常的胸腺或T淋巴细胞缺陷。
大样本研究显示临床表现为智力缺陷占92.3%,低钙占64.0%,腭异常占42.0%,心血管异常占25.8%。其他包括肥胖占35.0%,甲状腺功能低下占20.5%,听力缺陷占28.0%,胆石症占19.0%,脊柱侧弯占47.0%,皮肤异常(严重痤疮占23.0%、脂溢性皮炎占35.0%),精神分裂占22.6%[6]。
先天性无胸腺是DGA的标志特征,但完全性DGA仅占所有患者的不到1%[7]。临床表型同严重联合免疫缺陷病(SCID),预后恶劣。部分DGA更常见,免疫特征为部分联合免疫缺陷,临床表现为反复上呼吸道感染,下呼吸道感染少见。6月龄后出现荚膜菌引起的反复窦肺感染。伴T淋巴细胞减少者易于出现病毒,念珠菌感染或早期感染死亡,尤其伴CD4和CD8同时减少,胸腺输出减少或甲状旁腺减低者。
心脏异常主要包括累及流出道的各种异常,包括Fallot四联症、B型主动脉弓离断、永存动脉干、右主动脉弓及右锁骨下动脉畸形。甲状旁腺发育不良导致低钙,婴儿可出现手足抽搐或惊厥。由于胸腺不发育或发育不良导致T淋巴细胞缺陷,患者感染敏感性增高。
婴儿期可出现小下颌,低耳位伴垂直半径短和耳廓异常,内眦距过宽伴短的睑裂,斜视,人中短,小口,球形鼻,方鼻尖。由于黏膜下裂或腭裂致鼻音重。身材矮小。轻到中度学习困难。精神异常见于一小部分成人患者,包括偏执精神分类和抑郁症。少见特征包括甲状腺低下、唇裂和耳聋。
自身免疫见于各个年龄段,疾病随年龄不同而不同,如青少年类风湿关节炎,特发性血小板减少性紫癜,自身免疫性溶血性贫血,鱼鳞病,白化病,炎症性肠病,成人类风湿关节炎和风湿热。
神经系统可见骶脑脊膜膨出,脊柱裂,交通性脑积水,脊髓脊膜膨出,小头,胼胝体发育不良,小脑扁桃体下疝畸形。大脑影像异常包括外侧裂区多小脑回,程度不同,经常不对称,右侧明显。
精神系统可见侵略性爆发,冷漠,精神特征如妄想,幻觉,痴呆。
泌尿生殖系统可见单侧肾不发育,肾发育不良,肾盂积水,无输尿管,原发闭经,伴血性囊肿的处女膜闭锁。
眼部包括角膜后胚胎环,扭曲的视网膜血管,眼睑悬垂,斜视,上睑下垂,弱视,倾斜的视神经。巩膜角膜弹性层膨出,小眼球,眼前段发育异常,虹膜角膜黏连。
完全性DGA患者出生后严重T淋巴细胞减少(CD3+<0.05×109/L)。针对丝裂原的增殖反应缺失或极度减低。不典型完全性DGA婴儿可出现克隆性T淋巴细胞群,淋巴细胞数量及增殖功能可变,但原始CD4+T淋巴细胞缺乏[CD3+ CD45RA+CD62L+<0.05×109/L,或<5% CD3+T,或T细胞受体切除环(TRECs)<100/10 000 T淋巴细胞]。一些患者,B淋巴细胞减少是其特征之一,尤其婴儿期,随时间恢复正常。
完全性DGA患者IgG、IgA和IgM减低(尽管出生后数周内母体残留影响IgG)。部分型DGA患者抗体缺陷谱广泛,经常有轻到中度抗体受损,婴儿期明显。低IgG伴亚类缺陷亦有报道,很多患者最初低的免疫球蛋白会随年龄增长变为正常。针对多糖抗体反应缺陷较常见。
不典型完全性DGA婴儿可表现皮疹和淋巴结大,临床表现类似于伴嗜酸性粒细胞增多的SCID[9]。TCRVB呈单克隆性,同时具有无胸腺表型。
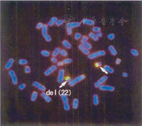
(1)婴儿期低钙是特征性表现,有时间段性,1年内可缓解。血甲状旁腺素(PTH)降低。(2)由于胸腺可位于其他位置或很小,不能凭外科手术、放射线或CT来诊断无胸腺,必须有分子生物学证据,如CD3+ CD45RA+ CD62L+ <0.05×10 9/L,或<5% CD3+T,或TRECs<100/10 000 T细胞。(3) 标准核型分析除外重要重组,或者单体型不平衡移位22pter-q11,或者染色体内缺失del(22)(q11.21q11.23)。用来源于缺失片段的探针行FISH[10],优先选择移位断裂点附近的探针(图1)。(4)如果没有细胞悬液或新鲜血液做核型分析,可用该区域一系列高变异探针来寻找位点缺失。目前常用的方法如多重连接探针扩增(MLPA)、比较基因组杂交(array-CGH)和拷贝数变异方法(CNV),见图2。

(1)补充钙剂和1,25胆骨化醇。(2)在免疫功能健全确认前,行外科手术需输注辐照的血,避免移植物抗宿主病(GVHD)。(3)完全性或不典型完全性DGA患者需立即转移至专业免疫中心行进一步评估和治疗[11]。启动抗卡氏肺囊虫肺炎、抗病毒、抗真菌的预防治疗和免疫球蛋白替代治疗。干细胞移植后可获得供者T淋巴细胞胸腺后的外周植入,但不能证明持续的T淋巴细胞生成。仅有数例长期存活报道,总存活率低(41%~48%),远低于其他SCID (80%)。原因主要为心脏外科情况[12]和GVHD。目前仅有2家实验室能够进行同种异体胸腺移植,存活率72%,致死的主要原因为感染。主要的远期不良反应是自身免疫病,如自身免疫性甲状腺疾病、1系、2系或3系血细胞减少,还包括肾病综合征和自身免疫性小肠炎。(4)部分性患者主要是对症治疗,随着年龄增长病情会减轻。细菌性窦肺感染需及时治疗。可能需要预防性抗生素,尤其冬季,有的患者可能需常年预防。伴有症状性低丙种球蛋白患者或预防效果不好的患者,可能需要丙种球蛋白替代治疗。活疫苗通常是安全的[13],建议CD4+T<0.4×109/L时避免接种活疫苗,由于保护性抗体水平维持时间短,应定期监测抗体水平,必要时重复接种疫苗。(5) 腭裂可能在黏膜下,需仔细寻找。环咽肌失功能需要尤其关注。语言治疗,教育辅助可能需要[14]。(6)成年患者需关注精神方面异常[15]。
22q11.2缺失综合征是最常见的染色体缺失综合征,由于咽弓发育异常,导致胸腺、甲状旁腺、心脏动脉圆锥和面容异常。完全性患者需要紧急救治,胸腺移植为治愈方法。部分性患者对症治疗,但心脏异常、环咽肌障碍影响预后。


























