
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Danon病是一种罕见的致死性X连锁显性遗传性疾病[1,2],曾被命名为X连锁的空泡性骨骼肌心肌病或酸性麦芽糖酶正常的溶酶体糖原贮积症,最早在1981年由Danon等[3]报道,在2000年由Nishino等[4]证实其致病基因是溶酶体相关膜蛋白2(LAMP2)基因。Danon病以男性多见,肥厚型心肌病、骨骼肌病和智力障碍三联征为其主要临床表现,病理特点为糖原颗粒堆积在心肌和骨骼肌细胞质内形成蜘蛛网样的膜性自噬溶酶体[5,6]。由于Danon病的罕见性,目前国内该病报道较少,现报道1例典型的Danon病,并分析其临床和病理以及基因改变特点。
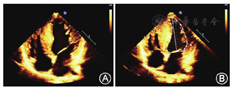
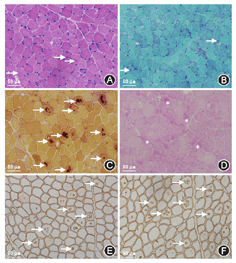
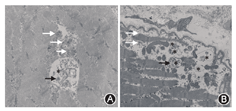
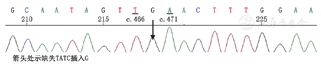
患者男性,15岁,因"发热伴头晕6 d"于2014年5月18日来我院就诊。患者于入院前6 d无明显诱因出现发热,体温最高达38.5 ℃,伴咳嗽咳痰,头晕,乏力,恶心呕吐,呕吐物为胃内容物,无胸痛、胸闷,无肩背部放射痛,无头痛,无意识丧失,就诊于当地医院,给予抗感染、抑酸等治疗,体温恢复正常,仍间断出现头晕、恶心呕吐。遂来我院就诊。患者自幼体弱,易感冒,体力较正常同龄者差,活动后易出现心悸、乏力、气喘,读书成绩不及格。入院时体检:智力低,发育差,身高160 cm,体重42 kg。双肺呼吸音粗,未闻及干、湿性啰音,心律齐,各瓣膜听诊区未闻及杂音,肝于肋下4 cm、剑突下5 cm可触及,脾肋下未触及。双上肢近端肌力Ⅴ级,双下肢近端肌力Ⅳ+级,远端肌力Ⅴ级。四肢腱反射减弱,病理征阴性。家族史:父母及姐姐体健,家族中其他成员无类似疾病。入院后主要生化检查:肌酸激酶2 626 U/L(正常值38~174 U/L),肌红蛋白1453 ng/ml(正常值0~85 ng/ml),肌酸激酶同工酶62.3U/L(正常值0~24.0 U/L),乳酸脱氢酶1 718 U/L(正常值120~300 U/L), ALT 1 043 U/L(正常值9~50 U/L),AST 882 U/L(正常值15~40 U/L)。抗核抗体、抗U1核糖核蛋白、抗Sm、抗SSA、抗SSB、抗Scl-70、抗Jo-1、抗r核糖核蛋白、抗双链DNA抗体均为阴性。类风湿因子、抗链球菌溶血素O、抗环瓜氨酸肽抗体呈阴性。肝炎筛查+戊型肝炎抗体测定结果为阴性。EB病毒、单纯疱疹病毒Ⅰ型及人巨细胞病毒核酸检测阴性。梅毒螺旋体抗体和人类免疫缺陷病毒抗体阴性。红细胞沉降率7 mm/h(正常值<15 mm/h),C反应蛋白13.1 mg/L(正常值0~8.0 mg/L),第三代促甲状腺素、总三碘甲状腺原氨酸、总甲状腺素、游离甲状腺素及游离三碘甲状腺原氨酸水平正常。辅助检查:胸部+全腹CT结果示:(1)全心增大,肺动脉增粗,宽径约3.5 cm;(2)两肺上叶可见多发斑片状密度增高影,以左肺上叶为主,边缘模糊考虑炎症;(3)心包积液,右侧胸腔少量积液;(4)肝脏增大,腹腔积液。心脏超声结果示:(1)室间隔非对称性肥厚(图1A),右心室壁及右心室节制索肥厚(图1B),考虑肥厚性心肌病;(2)二、三尖瓣轻中度关闭不全;(3)左心室正后壁基底段、中段及下壁基底段运动消失;(4)左心室收缩功能减低、舒张功能异常(Ⅲ度,限制性充盈),左心室充盈压增高;(5)右心室收缩功能及整体功能减低;(6)少量心包积液。心电图示:窦性心律,预激综合征图形,心率78次/min,Ⅰ、Ⅱ、Ⅲ、aVF、aVl、V1-6导联ST段压低,T波倒置。24 h动态心电图结果:窦性心律,平均心率为76次/min,最小心率55次/min,最大心率111次/min。室性早搏140个,呈单发、成对及室性心动过速。室上性早搏99个,呈单发、成对及房性心动过速。无心室停搏。ST-T段异常。肌电图示被检双胫前肌、右股三头肌、右三角肌呈肌源性损害。肱二头肌肌肉活体组织检查HE染色可见肌束内结缔组织无明显增生,肌间小血管壁无增厚,未见炎性细胞浸润。部分肌纤维胞质内有典型自噬空泡形成(图2A),胞质内可见不规则形状的嗜碱性物质沉积,肌纤维大小不等,直径20~50 μm,未见肌纤维变性,无核内移增加,未见不透明纤维和环状纤维。改良Gomori三色染色可见空泡样肌纤维(图2B),部分胞质内可见蓝紫色物质沉积。四氮唑还原酶染色和琥珀酸脱氢酶染色可见部分区域氧化酶缺失及中央轴空样肌纤维。非特异性酯酶染色可见空泡区域酶活性增强,部分肌纤维内可见棕黑色物质沉积(图2C)。酸性磷酸酶染色未见明显异常。油红O染色示个别肌纤维脂滴增多。过碘酸希夫染色部分肌纤维内阳性物质不均匀沉积(图2D)。ATP酶染色显示两型肌纤维呈镶嵌排列。免疫组织化学分析提示:肌纤维dystrophin(+),dysferlin(+),sarcoglycan(+)c-dystrophiny和α-sarcoglycan染色可见肌细胞内膜性自噬空泡形成(图2E、图2F)。电镜观察显示肌纤维内出现大量的膜性空泡,部分空泡内存在大量糖原颗粒、溶酶体颗粒以及自噬碎片,空泡周围有散在分布的糖原颗粒(图3A、图3B)。初步考虑为糖原贮积症、Danon病可能。蛋白质印迹证实lamp2蛋白缺失,送检肌肉标本进行LAMP2基因突变检测。检测结果:在LOVD数据库进行检索分析,送检标本LAMP2基因外显子4发现序列异常,c.467_470delTATCinsG,该改变尚无报道,属于移码突变(图4),该位点改变可导致编码氨基酸提前终止,正常人群中无此突变,临床意义明确,证实患者为Danon病。然而由于家属经济以及其他原因,未能进一步对患者基因进行家系验证,明确发病风险。追踪回访患者,令人遗憾的是患者出院仅半个月猝死,患者母亲偶感心悸,无其他不适。
Danon病是一种以肥厚型心肌病、骨骼肌病和智力障碍三联征为主要临床表现的X连锁显性遗传性疾病,以男性多见,多在20岁前出现心脏症状,并快速进展为晚期心力衰竭,常因猝死或心力衰竭而在30岁前死亡[2,7],女性较少发病。常表现为迟发或轻微的心脏症状。2000年Nishino等[4]首先发现LAMP2基因突变是Danon病的主要病因。
经典的Danon病临床表现无特异性,但除了上述其典型的临床三联征外,心电图以预激综合征最常见,还可伴有左心室高电压、房室传导阻滞、心房颤动、T波倒置、异常Q波等表现[8]。Arad等[9]认为,虽然Danon病引起的心肌肥厚似肥厚性心肌病,但二者有电生理差异的特征,尤以心室预激综合征可做鉴别。何继强等[10]的研究也支持了这一结果。Danon病最主要的病理学特点为肌细胞内可见不规则的膜性空泡以及糖原颗粒堆积的自噬溶酶体[6,11],而酸性麦芽糖酶正常,高碘酸希夫反应阳性。此外,Danon病可伴有血清肌酸激酶以及肝酶水平的升高[12,13]。本例男性患者自幼体弱,易患感冒,活动后易出现心悸、乏力、气喘,并且呈现出比较典型的肥厚型心肌病、骨骼肌病和智力障碍三联征的临床表现,心电图出现预激综合征,同时伴有血清肌酸激酶以及肝酶水平的升高,肌电图提示肌源性损害。于是我们给患者做了肌肉病理及电镜观察,结果显示患者肌细胞内膜性空泡并出现溶酶体自噬空泡[13,14],以及糖原颗粒的积聚。结合患者的临床表现,考虑为糖原贮积症、Danon病的可能。蛋白质印迹证实患者lamp2蛋白缺失,最终送检标本进行LAMP2基因检测,提示LAMP2基因外显子4发现序列异常(c.467_470delTATCinsG),属于移码突变,可导致编码氨基酸提前终止,临床意义明确,证实是Danon病。令人遗憾的是,我们未能对患者基因进行家系验证,明确其发病风险。Danon病目前仍无特异性的治疗方法,最有效的方法是心脏移植[15,16],可显著提高患者的生存率[13,17]。随着人们对Danon病的认识逐渐深入,相信未来一定可以寻找出针对Danon病的特异治疗方法。
总之,如果患者为年轻男性,自幼体弱,易出现心悸,检查发现存在心肌肥厚改变、骨骼肌病、智力障碍、血浆肌酸激酶水平升高,心电图发现预激综合征[7],应当高度警惕Danon病的可能,尽早进行LAMP2基因检测,从而确诊或除外Danon病。此外,对于确诊的Danon病患者以及其家属进行遗传咨询可以降低其后代的患病风险[18]。
无

























