
通过对3例红细胞丙酮酸激酶缺乏症(pyruvate kinase deficiency,PKD)患儿临床症状及PKLR基因新突变类型分析,探讨PKD的基因诊断方法。
应用目标序列捕获和高通量测序技术对3例PKD患儿的血液系统疾病相关基因进行测序,并预测基因突变对蛋白质功能的影响,确定患儿的致病基因型后,应用Sanger测序技术对此基因型进行验证。
在3例患儿中发现了5种PKLR基因突变类型,分别为c.1529G>A(p.R510Q)和c.1031T>G(p.I344S)复合杂合突变、c.847G>T(p.V283F)纯合突变,c.979delC(p.L327fs)和c.604_617del(p.V202fs)复合杂合突变,其中c.1529G>A(p.R510Q)突变已有文献报道,其余4种突变为PKLR基因新的突变类型。c.1031T﹥G(p.I344S)、c.847G﹥T(p.V283F)为可能致病变异,该基因突变致病的可能性>90%;c.979delC(p.L327fs)和c.604_617del(p.V202fs)变异为致病变异。且这5种突变经预测对蛋白质功能影响较大,均为致病突变。
对于临床诊断困难的PKD患儿,可通过目标序列捕获和高通量测序技术发现PKLR基因变异,并评价其致病性。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
红细胞丙酮酸激酶缺乏症(pyruvate kinase deficiency,PKD)是仅次于葡萄糖-6-磷酸脱氢酶(glucose-6-phosphate dehydrogenase,G-6-PD)缺乏症的最常见红细胞酶病,为常染色体隐性遗传病[1]。丙酮酸激酶(pyruvate kinase,PK)是糖酵解过程中关键限速酶之一,催化磷酸烯醇丙酮酸转变为丙酮酸[2,3],同时产生三磷酸腺苷(ATP),而成熟红细胞完全依赖于糖酵解途径供能,约50%的ATP来源于此步骤。PK缺乏导致红细胞内ATP生成减少,致使细胞膜钠钾泵和钙泵功能障碍,造成钾离子和水大量丢失,最终在脾、肝或骨髓内破坏而发生溶血。PK缺乏诊断较G-6-PD缺乏及其他酶缺乏诊断困难,许多患儿PK活力下降不明显,有些甚至明显高于正常活力测值,给PKD的诊断造成很大困难。鉴于此,基因诊断PKD的病例日益增多。本研究通过对上海交通大学医学院附属瑞金医院2015年1月至2017年3月收治的3例PKD患儿临床症状及PKLR基因新突变的报道,探讨PKD的PKLR基因诊断方法。现报告如下。
患儿1,男,5岁11个月时因"面色苍白5年余"就诊。患儿出生后即出现面色苍白,8个月时于当地医院就诊,提示贫血,输血1次后贫血好转,出院后未再正规诊治。患儿于5岁2个月就诊外院时血常规示血红蛋白(Hb) 66 g/L,胆红素增高,以未结合胆红素升高为主,尿胆原增高。骨髓细胞检查提示增生性贫血。患儿父母体健,无相关临床症状。体检:全身皮肤苍黄,睑结膜、口唇、甲床苍白,巩膜黄染,肝肋下3 cm,脾肋下3 cm。腹部B超:胆囊结石。住上海交通大学医学院附属瑞金医院期间输血治疗1次。该患儿出生后来院共输血2次。实验室检测结果见表1。
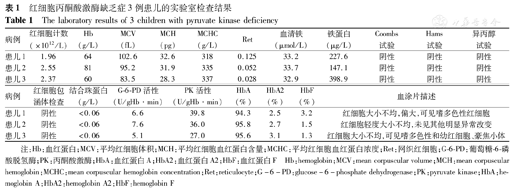
红细胞丙酮酸激酶缺乏症3例患儿的实验室检查结果
The laboratory results of 3 children with pyruvate kinase deficiency
红细胞丙酮酸激酶缺乏症3例患儿的实验室检查结果
The laboratory results of 3 children with pyruvate kinase deficiency
| 病例 | 红细胞计数(×1012/L) | Hb(g/L) | MCV(fL) | MCH(pg) | MCHC(g/L) | Ret | 血清铁(μmol/L) | 铁蛋白(μg/L) | Coombs试验 | Hams试验 | 异丙醇试验 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 患儿1 | 1.96 | 64 | 102.6 | 32.6 | 318 | 0.125 | 33.2 | 227.6 | 阴性 | 阴性 | 阴性 |
| 患儿2 | 2.55 | 81 | 95.2 | 31.9 | 335 | 0.052 | 33.7 | 147.1 | 阴性 | 阴性 | 阴性 |
| 患儿3 | 2.37 | 60 | 83.5 | 28.3 | 337 | 0.028 | 32.9 | 398.9 | 阴性 | 阴性 | 阴性 |
| 病例 | 红细胞包涵体检查 | 结合珠蛋白(g/L) | G-6-PD活性(U/gHb·min) | PK活性(U/gHb·min) | HbA (%) | HbA2 (%) | HbF (%) | 血涂片描述 |
|---|---|---|---|---|---|---|---|---|
| 患儿1 | 阴性 | <0.06 | 6.6 | 39.8 | 94.3 | 2.5 | 3.2 | 红细胞大小不均,偏大,可见嗜多色性红细胞 |
| 患儿2 | 阴性 | <0.06 | 7.6 | 36.0 | 95.8 | 2.7 | 1.5 | 红细胞轻度大小不均,未见其他明显异常改变 |
| 患儿3 | 阴性 | <0.06 | 5.1 | 27.0 | 95.6 | 3.1 | 1.3 | 红细胞大小不均,可见嗜多色性和幼红细胞、豪焦小体 |
注:Hb:血红蛋白;MCV:平均红细胞体积;MCH:平均红细胞血红蛋白含量;MCHC:平均红细胞血红蛋白浓度;Ret:网织红细胞;G-6-PD:葡萄糖-6-磷酸脱氢酶;PK:丙酮酸激酶;HbA:血红蛋白A;HbA2:血红蛋白A2;HbF:血红蛋白F Hb:hemoglobin;MCV:mean corpuscular volume;MCH:mean corpuscular hemoglobin;MCHC:mean corpuscular hemoglobin concentration;Ret:reticulocyte;G-6-PD:glucose-6-phosphate dehydrogenase;PK:pyruvate kinase;HbA:hemoglobin A;HbA2:hemoglobin A2;HbF:hemoglobin F
患儿2,女,7岁10个月时因"面色苍白7年余"就诊。患儿出生3 d出现新生儿高胆红素血症,予蓝光照射治疗。11个月时因皮肤、黏膜苍黄进行性加重,口唇指甲苍白就诊,血常规提示Hb 43 g/L,伴肝脾大,骨髓细胞检查提示增生性贫血骨髓象,予输血治疗后好转。2岁因"感冒"查血常规Hb 50 g/L,再次予输血1次。平时Hb为50~90 g/L,网织红细胞比例0.053~0.126。胆红素增高,以未结合胆红素升高为主,尿胆原增高。患儿父母体健,患儿弟弟出生后亦有严重黄疸,并出现胆红素脑病,有频繁抽搐病史,2岁时因抽搐时间过长死亡。体检:面色苍黄,巩膜黄染,口唇苍白,肢体扭转多动,语言发育迟缓,肝肋下3 cm,脾肋下6 cm。实验室检查结果见表1。
患儿3,男,出生2个月时因"发现贫血2个月"就诊,患儿胎龄36+5周因"胎儿宫内窘迫"剖宫产娩出,出生体质量1 560 g,出生Apgar评分9分,母孕史无特殊。患儿出生后(外院)红细胞计数1.87×1012/L,Hb 71 g/L,总胆红素144.8 μmol/L,结合胆红素18.9 μmol/L,未结合胆红素125.9 μmol/L,G-6-PD活性、Coombs试验均正常,予蓝光照射、输血等治疗。患儿父母体健,无相关临床症状,溶血试验未提示异常。患儿出生2个月就诊于上海交通大学医学院附属瑞金医院,皮肤轻度黄染,睑结膜、口唇、甲床苍白,肝肋下2 cm,脾肋下未触及,当时查Hb 60 g/L。给予输血治疗。目前平均每月输血1次。实验室检查结果见表1。
在知情同意的原则下,用乙二胺四乙酸抗凝管分别采集3例患儿及其父母外周血5 mL。按DNA提取试剂盒(北京迈基诺基因科技股份有限公司)说明书提取基因组DNA。本研究通过上海交通大学医学院附属瑞金医院医学伦理委员会批准[批号:(2017)临伦审第209号]。
首先将提取的基因组DNA超声波打断,并制备DNA文库,利用液相捕获试剂盒捕获血液系统疾病相关基因,对捕获到的序列进行PCR扩增,最后应用新一代测序仪illumina HiSeq2000进行高通量测序,寻找致病基因的突变。
对患儿及其父母外周血DNA进行Sanger测序,PCR及基因测序引物序列参照文献[3,4]设计。PCR扩增的反应体系(50 μL):DNA模板5 μL,10×PCR缓冲液5 μL,1×脱氧核糖核苷三磷酸5 μL,Taq酶2 U,上、下游引物各1.5 μL,补双蒸水至50 μL。反应条件:94 ℃ 30 s,55~65 ℃ 40 s,72 ℃ 45 s,共35个循环。将产物在15 g/L琼脂糖凝胶上进行电泳。DNA测序:PCR产物纯化后,用测序试剂盒在ABI 3730 DNA测序仪(美国Applied Biosystems公司)上进行序列测定。测序结果与GenBank已知的DNA序列(序列号:NG 011677.1)进行比对。
采用SIFT(sorting intolerant from tolerant)功能预测程序和PolyPhen-2软件预测点突变对PK蛋白功能的影响。
参照2015年美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)发布的《ACMG遗传变异分类标准与指南》[5]对遗传变异进行解读。
通过目标序列捕获高通量测序技术检测3例患儿血液系统疾病基因,发现PKLR基因编码区存在突变。应用Sanger测序法对患儿的突变位点进行验证,其结果与目标序列捕获高通量测序法完全一致。3例患儿基因检测结果如下:患儿1的PKLR基因编码区存在复合杂合突变,即第10外显子c.1529G>A(p.R510Q)和第7外显子c.1031T>G(p.I344S)(图1A、图1B)。这2种突变均为错义突变。患儿父亲携带第10外显子c.1529G>A(p.R510Q)杂合突变,患儿母亲携带第7外显子c.1031T>G(p.I344S)杂合突变,其中有文献报道了c.1529G>A(p.R510Q)突变[6]。这2种突变的SIFT功能预测结果分别为0.04和0,PolyPhen-2功能预测结果分别为0.993和1。表明这2种突变对PK蛋白功能的影响较大。患儿2的PKLR基因编码区存在纯合突变,即第6外显子c.847G>T(p.V283F)(图1C),该变异为错义突变。该变异在人群中发病率极低。患儿父亲和母亲该位点均为杂合变异。该突变的SIFT功能预测结果为0,PolyPhen-2功能预测结果为0.97。表明该突变对PK蛋白功能的影响较大。患儿3的PKLR基因编码区存在复合杂合突变,即第7外显子c.979delC(p.L327fs)和第5外显子c.604_617del(p.V202fs)(图1D、图1E),这2种突变均提前终止氨基酸链的编码,分别形成C末端缺失245和353个残基的截短型PK。患儿父亲携带第7外显子c.979delC(p.L327fs)杂合突变,患儿母亲携带第5外显子c.604_617del(p.V202fs)杂合突变。
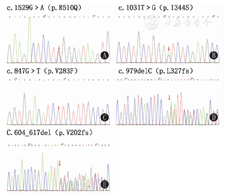
The Sanger sequen-cing of the <i>PKLR</i> gene in 3 patients with pyruvate kinase deficiency
注:A、B:患儿1;C:患儿2;D、E:患儿3 A,B;patient 1;C:patient 2;D,E:patient 3
The Sanger sequen-cing of the <i>PKLR</i> gene in 3 patients with pyruvate kinase deficiency
参照《ACMG遗传变异分类标准与指南》[5]对本研究中3例患儿的遗传变异进行解读,结果见表2。患儿1的c.1031T>G(p.I344S)突变,经检索千人数据库及EXAC数据库发现该突变属于隐性遗传病中极低频位点,符合中等致病证据(moderatepathogenicity,PM)2;其反式位置上检测到的变异c.1529G>A(p.R510Q)为已经明确报道过的致病变异[6],故符合PM3;经SIFT功能预测程序和PolyPhen-2软件预测c.1031T>G(p.I344S)变异对蛋白功能有较大影响,符合支持致病证据(supportingpathogenicity,PP)3;另外患儿1的临床表现高度符合PKLR基因突变所致表型,故满足PP4。因此c.1031T>G(p.I344S)为可能致病变异,该基因突变致病的可能性>90%。患儿2的c.847G>T(p.V283F)突变经检索千人数据库及EXAC数据库发现该突变属于隐性遗传病中极低频位点,符合PM2;c.847G>T(p.V283F)突变导致氨基酸的变化之前未曾报道过,但是在同一位点,导致另外一种氨基酸的变异已经确认是致病性的[7],故符合PM5;经SIFT功能预测程序和PolyPhen-2软件预测c.847G>T(p.V283F)变异对蛋白功能有较大影响,符合PP3;另外患儿2的临床表现高度符合PKLR基因突变所致表型,故满足PP4。因此c.847G>T(p.V283F)亦为可能致病变异。患儿3的c.979delC(p.L327fs)和c.604 617del(p.V202fs)突变为移码突变,均符合非常强的致病性证据1(very strongpathogenicity1,PVS1);经检索千人数据库及EXAC数据库发现这2种突变均属于隐性遗传病中极低频位点,符合PM2;这2种突变均导致碱基缺失,符合PM4;移码突变c.979delC(p.L327fs)形成C末端缺失245个残基的截短型PK,对蛋白功能有较大影响,移码突变c.604_617del(p.V202fs)形成C末端缺失353个残基的截短型PK,对蛋白功能有较大影响,均符合PP3;另外患儿3的临床表现高度符合PKLR基因突变所致表型,故满足PP4。因此患儿3的c.979delC(p.L327fs)和c.604_617del(p.V202fs)变异为致病变异。
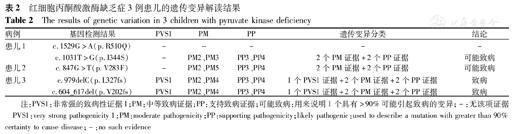
红细胞丙酮酸激酶缺乏症3例患儿的遗传变异解读结果
The results of genetic variation in 3 children with pyruvate kinase deficiency
红细胞丙酮酸激酶缺乏症3例患儿的遗传变异解读结果
The results of genetic variation in 3 children with pyruvate kinase deficiency
| 病例 | 基因检测结果 | PVS1 | PM | PP | 遗传变异分类 | 结论 |
|---|---|---|---|---|---|---|
| 患儿1 | c.1529G>A(p.R510Q) | - | - | - | - | - |
| c.1031T>G(p.I344S) | - | PM2、PM3 | PP3、PP4 | 2个PM证据+2个PP证据 | 可能致病 | |
| 患儿2 | c.847G>T(p.V283F) | - | PM2、PM5 | PP3、PP4 | 2个PM证据+2个PP证据 | 可能致病 |
| 患儿3 | c.979delC(p.L327fs) | PVS1 | PM2、PM4 | PP3、PP4 | 1个PVS1证据+2个PM证据+2个PP证据 | 致病 |
| c.604_617del(p.V202fs) | PVS1 | PM2、PM4 | PP3、PP4 | 1个PVS1证据+2个PM证据+2个PP证据 | 致病 |
注:PVS1:非常强的致病性证据1;PM:中等致病证据;PP:支持致病证据;可能致病:用来说明1个具有>90%可能引起致病的变异;-:无该项证据 PVS1:very strong pathogenicity 1;PM:moderate pathogenicity;PP:supporting pathogenicity;likely pathogenic:used to describe a mutation with greater than 90% certainty to cause disease;-:no such evidence
PKD是一种常见的遗传性红细胞酶病。红细胞酶活性的测定是诊断红细胞酶病的金标准,通过测定酶活性的方法可使大多数患者明确诊断[8]。然而还有一部分PKD患者PK活性下降不明显甚至增高,其确切机制尚不明确,可能与下列因素有关:PK是年龄依赖性酶,即细胞年龄越轻,酶活力越高;外周血网织红细胞计数增高,亦会引起酶活力代偿性增高;溶血发作期骨髓红系增生活跃,大量新生红细胞进入循环也使得酶活力升高;近期输血亦可掩盖酶缺陷。因此,评价PK活力应注意网织红细胞计数,并建议溶血发作或输血3个月后复查酶活力。此外,PK同工酶中白细胞M2型PK(PKM2)活性较红细胞R型PK(PKR)高300倍左右,试验中白细胞去除不彻底亦可能明显影响活性的测定值;PKM2亦存在于有核红细胞中,溶血严重时外周血中的晚幼红细胞亦可影响测定值。上述诸多因素均可影响PK活性的测定,给PKD的确诊带来了很大的困难。随着基因诊断技术的发展,PKD也可通过测定PKLR基因的外显子、侧翼区及启动子序列等明确诊断。基因诊断适用于无法通过常规实验室检查方法确诊的PKD病例。本研究中的3例患儿的病史和检查结果提示可能为溶血性贫血,但溶血试验检查为阴性,PK活性亦正常,最终,通过基因诊断技术检测到患儿PKLR基因存在致病突变,确定了PKD的诊断。
PK由PKLR和PKM 2种基因编码的4种同工酶(L、R、M1、M2)组成,其中PKLR基因编码PKL和PKR 2种同工酶,PKR主要存在于成熟红细胞,PKL主要分布在肝脏、肾皮质和小肠[9]。PKM基因编码M1型PK和PKM2 2种同工酶,PKM1分布于骨骼肌、脑,PKM2分布于白细胞、血小板、胎儿组织、肺、脾、肾、脂肪组织及有核红细胞[10,11]。PKLR基因突变可导致红细胞PKR变异,从而引起PKD。
PKLR基因位于染色体1q21,包含12个外显子。迄今为止,已报道的PKLR突变类型已有250余种,包括错义突变、无义突变、碱基缺失、碱基插入等[12]。其中多数为基因编码区的点突变,美国、北欧和欧洲中部地区以1529A最为常见,欧洲南部地区以1456T常见,而亚洲以1468T最为常见[6,13,14,15,16]。本研究应用目标序列捕获高通量测序结合Sanger测序技术检测到3例患儿的PKLR基因编码区存在5种突变。检索国内外相关文献,迄今为止,除c.1529G>A(p.R510Q)突变外,本研究检测到的其余4种突变类型的报道尚属首次。因此对于临床诊断困难的病例,可通过目标序列捕获高通量测序结合Sanger测序技术进行PKD基因诊断及PKLR基因新突变类型的发现。
PKD患者临床表现严重程度不一,部分患儿在新生儿期即发生高胆红素血症,以致需要进行光疗,甚至换血治疗,有的患者临床表现比较轻微,但在感染、劳累等情况下贫血症状可加重。有临床症状的PKD患者多为复合杂合子或纯合子[8]。结合3例患儿的临床表现及基因检测结果发现,3例患儿临床表现是依次加重的,不同基因位点的复合杂合突变临床表现较轻,同一位点的纯合突变次之,移码突变的复合杂合突变的临床表现最重。因此,对于临床诊断困难的病例,不仅可通过基因诊断确诊PKD,且可预测患者临床表现的严重程度。

























