
Cockayne综合征是一种罕见的常染色体隐性遗传病,由DNA修复障碍而导致多系统退行性损害,主要表现为发育落后、生长迟缓、早衰、光敏感及小头畸形。Cockayne综合征的临床表型为一个连续而重叠的谱系,从重到轻依次为脑-眼-面-骨综合征(COFS)、Cockayne综合征Ⅱ型、Cockayne综合征Ⅰ型、Cockayne综合征Ⅲ型及紫外线敏感综合征;另外还有着色性干皮病-Cockayne综合征型。Cockayne综合征在细胞水平的表现为紫外线照射后DNA修复缺陷,已知主要致病基因为CSA(ERCC8)与CSB(ERCC6)。现拟就Cockayne综合征的临床及遗传学研究进展进行综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Cockayne综合征是一种罕见的常染色体隐性遗传病,以发育落后、生长迟缓、早衰、光敏感及小头畸形为主要特征,常伴多系统损害。Cockayne[1]在1936年首次报道了1例"侏儒伴视网膜萎缩和耳聋"的患儿,CSA(ERCC8)与CSB(ERCC6)分别在1992年、1995年被确定为Cockayne综合征的主要致病基因[2,3]。早发型(Ⅱ型)、晚发型(Ⅲ型)与最初描述的经典型具有相同的细胞学缺陷[4]。Nance和Berry[5]在1992年建立了Cockayne综合征的诊断标准。Cockayne综合征表型为一个连续而重叠的谱系,从重到轻依次为脑-眼-面-骨综合征(COFS)、Cockayne综合征Ⅱ型、Ⅰ型、Ⅲ型及紫外线敏感综合征[6]。另外,极少数患者同时具有Cockayne综合征和着色性干皮病表型,称为着色性干皮病-Cockayne综合征型[7]。国外资料显示,日本Cockayne综合征的发病率为1/277万,西欧为1/270万[8,9],我国尚无统计学报道。现就Cockayne综合征的临床与遗传学研究进展进行综述。
COFS为Cockayne综合征变异型,是最严重的亚型,最初在曼尼托巴土著居民中被报道,主要表现为关节屈曲、小头畸形、白内障及小眼畸形[10]。
又称重型或早发型,出生后即出现生长迟缓,可伴先天性白内障或眼部其他结构性病变,在儿童早期即可出现脊柱和关节的挛缩,常在7岁前死亡[11]。
又称经典型或温和型,胎儿期正常,2岁以内出现生长发育异常,身高、体质量及头围均远远小于健康同龄儿第50百分位数,伴进行性视听损害、神经系统损害,常在20岁内死亡[12]。
为轻型或晚发型,生长和认知的发育基本正常,或后期出现发育落后[13]。
为Cockayne综合征变异型,仅表现为皮肤光敏感、雀斑、肺血管扩张及皮肤干燥,无其他临床特点,亦无肿瘤发生倾向[14]。
根据基因型分为不同的亚型,临床特点包括着色性干皮病典型的面部雀斑和早期皮肤癌,及Cockayne综合征的临床特征,如智力损害、癫痫、体格落后及性腺功能低下[7]。
眼球凹陷是Cockayne综合征的常见特征之一,由皮下和眼眶脂肪减少所致,在疾病早期不易识别。脑-眼-面-骨综合征主要表现为小下颌、低耳位,部分患者还有小眼球[5]。
生长迟缓及早衰是Cockayne综合征突出且持续存在的特征,也是疾病最早期的表现之一。Cockayne综合征Ⅰ型和Cockayne综合征Ⅱ型患者体质量比身高更早地受到影响,并且受累程度更严重,而轻度受累患者身材矮小是最主要的表现。Cockayne综合征Ⅰ型和Cockayne综合征Ⅲ型患者出生时的体质量和身长往往正常,Cockayne综合征Ⅰ型在1岁以后、Cockayne综合征Ⅲ型2岁以后往往可观察到生长速度减慢,5岁以后常有早老症样表现。Cockayne综合征Ⅱ型在婴儿期、Cockayne综合征Ⅰ型在儿童期、Cockayne综合征Ⅲ型在青少年晚期分别出现体格发育减缓或停滞,所有Cockayne综合征患儿的体质量和身高最终均<-3s[15]。
发育落后是Cockayne综合征的主要表现,在疾病后期所有亚型均可能发生神经系统退化和认知衰退。对于COFS和Cockayne综合征Ⅱ型患者,通常在新生儿期存在喂养困难和哭声弱,并伴轴向肌张力低下和外周性肌张力增高,通常不能独坐或独站,无语言表达或仅能说简单词语。Cockayne综合征Ⅰ型患儿出生后数月可暂无明显发育异常,通常可学习走路并能理解简单词语,但2岁左右开始出现运动和语言倒退[1]。Cockayne综合征Ⅲ型患者在学龄期可存在轻微的智力障碍,通常在30~40岁以后出现认知衰退和早期痴呆。
大多数Cockayne综合征患者显示锥体、锥体外系、小脑及外周神经损害,并随年龄增长日益加重。小头畸形为其主要临床特征,COFS和Cockayne综合征Ⅱ型患者出生时即可出现头围偏小,3岁以前无小头畸形为Cockayne综合征Ⅰ型排除标准[5]。小脑共济失调多见于晚发型患者,癫痫发生率高于一般人群。
随疾病进展患儿大小脑萎缩、颅内钙化和白质异常是Cockayne综合征的主要神经影像学特征[16]。白质减少和脑室扩大是最早可检测到的异常,颅内钙化可见于脑基底核、齿状核及皮层下白质[17],但1岁内可不明显。密集或点状、对称性壳核钙化主要见于Cockayne综合征Ⅰ型和Cockayne综合征Ⅲ型患者,并与后期齿状核钙化相关;弥散性皮质下钙化主要见于Cockayne综合征Ⅱ型和COFS患者。
关节炎和先天性脊柱后凸是COFS特有的临床特征,需要早期矫形治疗。
大多数Cockayne综合征患者可出现牙釉质发育不良,50%~75%的患者存在龋齿[5]。
皮肤光敏感是Cockayne综合征的次要诊断标准,为2/3~3/4患者的突出临床特点。患者短时间暴露于阳光,即可发生不同程度晒伤,年长患者可见色素斑。其他皮肤损害包括少汗、头发干而细、指甲营养不良等[18]。少数着色性干皮病-Cockayne综合征型患者还可并皮肤恶性肿瘤。
进行性感音神经性耳聋是Cockayne综合征的常见表现,在病程早期并不明显。尽管Cockayne综合征Ⅰ型患者常伴智力损害,但助听器依然是有益的,已有数例耳蜗植入病例报道[19]。Cockayne综合征Ⅲ型患者听力损害较轻,常在病程晚期出现。
进行性视网膜色素变性是Cockayne综合征的主要特征之一,在疾病晚期显著,眼底检查常显示经典的"椒盐样"视网膜病变。COFS等严重患者也可出现白内障及眼睛结构异常,包括小眼畸形和虹膜发育不全等。
Cockayne综合征患者肾损害可表现为慢性或急性,较常见的有慢性高血压、中度蛋白尿、高尿酸血症,可导致进行性肾衰竭。在疾病晚期急性肾病、肾衰竭是导致死亡的原因之一[9]。
Cockayne综合征患者常并肝损害,不伴凝血因子异常,偶尔有肝大,少数患者并无症状性胆汁淤积[5]。
动脉粥样硬化是Cockayne综合征患者常见的病理表现,可累及脑、冠状动脉和主动脉[15]。
患者常见高胰岛素血症和糖耐量异常,一些青年Cockayne综合征Ⅲ型患者并糖尿病。胰岛素样生长因子-1和甲状腺激素水平通常在正常范围内[5]。
绝大多数Cockayne综合征青春期可有不同程度性发育。有文献报道数例轻型女性患者怀孕,但由于母亲身材矮小,易引起流产或早产[20]。男性患者常有隐睾和生殖器不发育。
皮下脂肪减少、进行性听力损害、认知障碍、肾功能衰退、动脉粥样硬化、慢性高血压及糖尿病为Cockayne综合征的临床特征,类似于正常衰老。
患者个体差异显著,症状复杂,不同亚型发病时间和进展速度不同。COFS、Cockayne综合征Ⅱ型、Cockayne综合征Ⅰ型、Cockayne综合征Ⅲ型及紫外线敏感综合征临床表现是连续而重叠的,因此需要具体的诊断标准予以鉴别,但总体来说发病越早的患儿病情越重、寿命越短。
COFS表型最重,关节炎是一个重要的特征,可作为诊断要点,其他表现与Cockayne综合征Ⅱ型重叠。需要注意的是Cockayne综合征Ⅱ型患者通常在出生时出现不同程度的外周性肌张力增高和进行性关节挛缩,但在COFS患者,神经损害较Cockayne综合征Ⅱ型出现的早。
最轻型的紫外线敏感综合征由于临床症状较轻微,确诊也存在一定困难,目前仅有极少数成人发病的报道,均由CSB突变所致。紫外线敏感综合征的显著特点为智力正常,除极少数患者身材矮小和皮肤光敏感外,在成人期之前无任何临床症状。
髓鞘形成低下、钙化和脑萎缩为主要特征。钙化常发生在壳核,少数在额顶叶皮质。幕上白质、小脑、胼胝体和脑干可见严重的进行性萎缩[16]。
CSB、CSA及UVSSA缺陷可导致转录偶联修复通路(transcription-coupled repair,TCR)缺陷,可通过损伤后RNA合成修复(recovery of RNA synthesis,RRS)进行检测。ERCC1、XPB、XPD、XPG及XPF缺陷导致全基因组修复(global genome repair,GGR)和TCR共同缺陷。GCR缺陷可通过非程序化DNA合成(Unscheduled DNA synthesis,UDS)进行检测[21]。
Cockayne综合征的主要致病基因为CSB和CSA,其中CSB突变约占65%,CSA突变约为35%[22]。已报道97种CSB突变和43种CSA突变与Cockayne综合征相关(Human Gene Mutation Database)。CSB编码的DNA剪切修复蛋白ERCC6主要参与紫外线照射诱发的DNA损伤修复。CSA编码高度保守的WD40重复序列蛋白[23]。
XPB(ERCC3)、XPD(ERCC2)、XPF(ERCC4)和XPG(ERCC5)突变的Cockayne综合征患者具有严重的光敏性及皮肤癌风险,表现为严重的体格和神经损害。COFS是Cockayne综合征最严重的亚型,由ERCC1、XPD、XPG及CSB突变致病。着色性干皮病-Cockayne综合征型主要由XPB、XPD、XPF及XPG基因缺陷致病。紫外线敏感综合征由CSA、CSB及UVSSA突变致病。
因为CSB、CSA编码的蛋白在转录偶联的核苷酸切除修复中起重要作用,所以该基因缺陷易导致Cockayne综合征患者的皮肤光敏感,但患者同时出现生长迟缓和神经退行性变的具体分子机制尚不清楚[24]。
早期对CSA、CSB与Cockayne综合征亚型的研究未发现显著的基因型-表型相关性。
1992年由Nance和Berry[5]定义了最初的Cockayne综合征临床诊断标准,当时尚未具备成熟的分子遗传学诊断技术。2013年Laugel[6]对临床诊断标准进行了修订(表1),对此病诊断的特异性和敏感性均分别达到98%、90%,阳性和阴性预测值为97%。
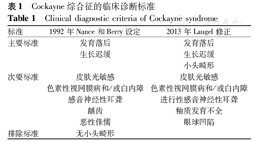
Cockayne综合征的临床诊断标准
Clinical diagnostic criteria of Cockayne syndrome
Cockayne综合征的临床诊断标准
Clinical diagnostic criteria of Cockayne syndrome
| 标准 | 1992年Nance和Berry设定 | 2013年Laugel修正 |
|---|---|---|
| 主要标准 | 发育落后 | 发育落后 |
| 生长迟缓 | 生长迟缓 | |
| 小头畸形 | ||
| 次要标准 | 皮肤光敏感 | 皮肤光敏感 |
| 色素性视网膜病和/或白内障 | 色素性视网膜病和/或白内障 | |
| 感音神经性耳聋 | 进行性感音神经性耳聋 | |
| 龋齿 | 釉质发育不全 | |
| 恶性侏儒 | 眼球凹陷 | |
| 排除标准 | 无小头畸形 |
但修正后的标准仍只适用于狭义的Cockayne综合征,即Cockayne综合征Ⅰ~Cockayne综合征Ⅲ型,而不适用于COFS和紫外线敏感综合征。另外,在疾病早期即使只符合2个主要标准也要考虑Cockayne综合征的可能。
COFS的主要诊断标准:先天性小头畸形、先天性白内障和/或小眼畸形、关节挛缩、重度发育落后、出生后重度生长迟缓、面部异常及TCR缺陷[10]。紫外线敏感综合征诊断要点:体格发育落后或光敏感,伴进行性智力下降、共济失调或听力损害,通过DNA修复检测可进一步确定。
目前,主要运用二代测序技术进行DNA或RNA测序分析进行基因诊断[25]。皮肤成纤维细胞紫外线照射后RNA合成修复缺陷是Cockayne综合征确诊的金标准。
当患者出现以下异常时,需要考虑鉴别诊断,如:面部、四肢、心脏及内脏的先天性异常,反复感染,代谢或神经危象,血液学异常,各种肿瘤。
生长迟缓可见于内分泌、代谢或胃肠道疾病、染色体病和一些临床综合征;大部分白质脑病患者不伴生长迟缓;脑内钙化可能提示先天性感染或钙磷代谢障碍及Aicardi-Goutieres综合征;皮肤薄、光敏感,头发稀少症状突出,需考虑着色性干皮病、Bloom综合征及其他早老症;早发色素性视网膜病可能提示线粒体病或过氧化物酶体缺陷[26];Warburg Micro综合征与Cockayne综合征Ⅱ型、COFS临床表现类似,但无快速进展性神经退行性变,DNA修复功能正常。
目前主要是对症支持治疗,国外曾认为高脂饮食对于延缓早衰进展有一定效果,但对皮肤、脑、肝、骨骼等器官损害效果不明显[27]。
所有Cockayne综合征亚型均符合常染色体隐性遗传病特征,针对基因诊断明确的家庭,在母亲再次妊娠时可通过羊水细胞或胎盘绒毛细胞基因分析进行胎儿出生前诊断。另外,还可通过羊水细胞DNA修复功能试验进行产前诊断[28]。植入前诊断理论上讲是可行的,但在Cockayne综合征家庭未见报道过。

























