
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿男,5岁9月龄,汉族。因"反复发热伴面色苍白5年余"于2017年5月入住深圳市儿童医院风湿免疫科。患儿5年余前(生后10 d左右)出现反复发热,以中高热为主,热峰39 ℃以上,间隔2~3个月发作1次,每次持续3~4 d,3年前(2岁龄)发热频次增多,间隔2~4周发作1次,每次持续3~7 d,发热时伴游走性关节肿痛及活动障碍,以双膝、双踝、双肘、双腕关节为主,伴脐周剧烈腹痛、呕吐,间断腹泻,偶有头痛,持续性面色苍白,外院曾诊断为"脓毒症、川崎病、慢性贫血、反应性关节炎、幼年特发性关节炎"等,予抗感染、补铁、非甾体类抗炎药治疗后病情仍反复,2年前临床诊断"急性化脓性阑尾炎",行腹腔镜阑尾切除术,2个月前曾诊断"不完全性低位肠梗阻",行禁食、胃肠减压等对症支持治疗后好转。5 d前患儿再次出现发热,伴颜面、双手红斑样皮疹,无痒感,伴腹痛及关节肿痛,以"自身炎症性疾病"收入院。个人史、家族史无特殊。
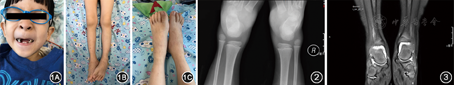
体格检查:身高94 cm,体重18.5 kg。神志清,反应可,体形消瘦,面色苍白,全身无皮疹;颈部、颌下及锁骨上可扪及肿大淋巴结,最大2 cm×1.5 cm,质软光滑,无触痛,活动度欠佳,局部无红肿;口唇干燥、苍白,可见龋齿,呼吸稍促,双肺呼吸音粗,未及干湿性啰音;心音有力,律齐,未闻及病理性杂音;腹部膨隆,肝肋下4 cm,质韧光滑,脾脏右肋下平髂前下棘。双膝、双踝、右腕关节肿胀伴活动受限(图1),局部皮温升高,触痛明显,四肢肌力及肌张力正常,神经系统查体未见异常。毛细血管充盈时间1 s。
实验室检查:发热时血常规:白细胞(5.13~10.81)×109/L[参考值(5~12)×109/L],中性粒细胞百分比0.566~0.787(参考值0.500~0.700),C反应蛋白(CRP)51.2~105.9 mg/L(参考值0~10 mg/L);体温正常时血常规:白细胞(6.35~10.63)×109/L,中性粒细胞百分比0.691~0.762,CRP 8.6~17.2 mg/L。血红蛋白(65~102)g/L(参考值110~160 g/L),红细胞体积(72.1~78)fl(参考值82~96 fl),平均红细胞血红蛋白含量(22.4~25.4)pg(参考值27~32 pg),网织红细胞比例0.65%(参考值0.45%~1.36%),网织红细胞绝对值23.30×109/L[参考值(17.0~70.1)×109/L];直接抗人球蛋白试验(+),间接抗人球蛋白试验(-);叶酸、维生素B12、铁蛋白无异常;降钙素原(0.12~0.53)μg/L(参考值0~0.5 μg/L);红细胞沉降率(51~122)mm/1 h(参考值0~15 mm/1h);血清淀粉样蛋白(SAA)(127~158)mg/L(参考值0~6 mg/L);血生化:球蛋白(38.1~52.8)g/L(参考值20~30 g/L),总胆固醇(2.07~2.97)mmol/L(参考值3.1~5.8 mmol/L)。体液免疫:IgA 5.31~11.26 g/L(参考值0.41~2.97 g/L),IgM、IgG、补体C3、补体C4大致正常。骨髓涂片示骨髓增生明显活跃。粪尿常规、肝酶、肾功能、心肌酶、电解质、类风湿因子、肿瘤标志物、肾小管标志物、葡萄糖-6-磷酸脱氢酶活性、地中海贫血基因筛查、淋巴细胞免疫分型、自身抗体谱、血液串联质谱、尿气相质谱均未见异常。常见病原学检查无异常,包括血培养、结核免疫分析、EB病毒抗体与DNA、围产儿畸形病原体检测、抗链球菌溶血素"O"试验和肥达试验等。
影像学检查:腹部超声提示肝脾弥漫性增大(肝右肋下3 cm、脾左肋下4 cm),肠系膜多增大淋巴结;关节超声提示双肘、双膝、双踝、双腕关节滑膜囊积液伴滑膜增厚;踝关节正侧位X线片提示双踝关节软组织肿胀,关节面光滑,关节对位良好(图2);踝关节磁共振成像(MRI)提示双踝关节积液,滑膜明显增厚伴强化(图3)。心电图、脑电图、心脏超声、胸腹CT、头颅MRI均未见明显异常。
基因检测:患儿甲羟戊酸激酶(MVK)基因存在2个杂合变异:(1).c.827A>G,p.E276G,为错义变异,经家系验证分析提示患儿父亲为该位点杂合变异,患儿母亲该位点无变异;(2).c.1127G>A,p.G376D,为错义变异,经家系验证分析提示患儿母亲该位点杂合变异,患儿父亲该位点无变异(图4)。
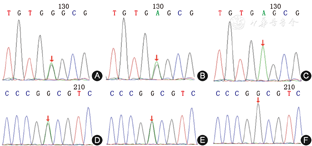
箭头所指为变异相关位点
出院诊断:甲羟戊酸激酶缺乏症(MKD),溶血性贫血,机械性肠梗阻,中度脱水。
诊治经过:本例患儿符合高IgD伴周期性发热综合征(HIDS)的临床特征,为进一步验证HIDS诊断,采集40例健康儿童及HIDS患儿血清测定MVK与IgD水平,并建立正常参照值,HIDS患儿急性发作期与发作间歇期的MVK分别为10.987 μg/L与25.078 μg/L,均显著低于正常参照值(96.17±19.24)μg/L;而发作期与间歇期的血清IgD分别为4 894.97 mg/L和4 968.75 mg/L,均显著高于正常参照值(44.11±28.51)mg/L。患儿MVK基因的2个变异位点在人类基因突变数据库(HGMD)中均未见报道,不属于多态性位点,在人群中发生频率极低,功能验证显示MVK活性显著降低,故该复合杂合变异确定为致病性变异。治疗上予布洛芬对症退热效果欠佳,甲泼尼龙可控制症状,禁食、胃肠减压、静脉营养等对症支持治疗后病情好转出院。
出院随访:出院后长期口服泼尼松片5~10 mg/d,患儿发热、腹痛及关节症状控制尚可,贫血明显改善,可正常上学。
本病例资料和涉及研究的内容已获深圳市儿童医院伦理委员会批准(2013-07-24函和2016-021函),患儿监护人已签署知情同意书。
MKD是一种罕见的自身炎症性疾病,属常染色体隐性遗传病,致病基因位于染色体12q24,所编码的MVK是甲羟戊酸途径的关键酶,目前根据MVK酶活性缺陷程度分为HIDS和甲羟戊酸尿症(MA)两种表型[1,2],本例患儿为HIDS表型。总结国外多中心大样本资料[3,4,5,6],MKD的确诊存在明显的滞后性,多数患儿确诊前曾被误诊为家族性地中海热、幼年特发性关节炎、风湿热、反复或慢性感染、白塞病等,本例患儿新生儿期起病,历经5年余才得以确诊。MKD患儿多在1岁前起病,诱因包括疫苗接种、感染和应激等,80%以上患儿在发作间歇期可无症状。几乎所有HIDS患儿均有发热症状,持续3~7 d,半数以上患儿发热时伴腹痛、腹泻、呕吐、淋巴结肿痛、皮疹、头痛、关节痛、肌痛和口腔溃疡,皮疹和关节症状可持续存在,其他症状多随热退而缓解。神经系统受累可出现小脑综合征、癫痫和精神发育迟滞,部分可出现色素性视网膜炎、葡萄膜炎、视野缺损、白内障,少数可并发巨噬细胞活化综合征。远期并发症主要为内脏淀粉样变性、肠黏连以及关节挛缩等。相较于HIDS,MA起病更早,神经系统受累更重,可出现宫内发育迟缓、先天畸形甚至死产或早夭[1],多伴有特殊面容,如前额隆起、眼距增宽、睫毛过长以及三角面容等,可有胆汁淤积和肝功能异常。急性发作期多伴非特异性炎症指标升高。典型的MKD患儿有多伴血清IgD升高和(或)血清IgA水平升高,多数患儿有尿甲羟戊酸排泄增加,目前认为MVK水平降低有确诊意义。本例患儿于新生儿期起病,以周期性反复性发热、剧烈腹痛、呕吐、关节痛、头痛为主要表现,急性发作期存在非特异性炎症指标升高,同时伴有血清MVK水平降低、血清IgD、IgA升高,故符合HIDS临床特征。2015年欧洲儿童风湿病组织制定了MKD的临床诊断标准,即具有该病的典型的反复发作性发热(除外感染及其他原因),并有发病年龄小于2岁(10分)、口疮性口炎(11分)、淋巴结肿大或脾肿大(8分)、淋巴结疼痛(13分)、阵发性腹泻(20分)、持续性腹泻(37分)以及不合并胸痛(11分),如果切值≥42分即可诊断,该诊断标准的特异度为89%,灵敏度为53%[7]。根据上述诊断标准,本例患儿总分49分(起病年龄小于2岁、肝脾淋巴结肿大、腹泻、无胸痛),且存在周期性发热,辅助检查提示MVK水平显著降低、血清IgD和IgA水平显著升高、急性期非特异炎症指标明显升高等,基因检测提示MVK基因复合杂合变异,且排除了感染及肿瘤性疾病,因此HIDS诊断成立。本例患儿存在自身免疫性溶血性贫血(AIHA),目前HIDS合并AIHA罕有报道,考虑与长期的自身炎症性疾病以及脂质代谢异常导致的适应性免疫功能紊乱有关[8,9]。
MKD尚无根治方法,遵循个体化治疗原则,以对症支持治疗为主,首选非甾体类抗炎药,二线治疗包括糖皮质激素及生物制剂[10],本例患儿对非甾体类抗炎药疗效欠佳,给予糖皮质激素口服可控制症状,并正常上学。造血干细胞移植成功病例亦有报道[11],植物类异戊二烯可能成为治疗MKD的新靶点。

























