
为了筛查神经纤维瘤病1型(NF1)一遗传家系致病基因突变位置及分析其临床表型,笔者用Illumina Miseq测序对NF1一家系的先证者进行了目标区域捕获及分析,寻找可疑突变位点,并行家系成员Sanger测序验证。结果显示,先证者携带了2个罕见变异,除KIF1B基因c.C3649T杂合错义变异(KIF1B: p.P1217S het)外,还发现了NF1基因(NM_000267.3)外显子41的c.T6311C(p.L2104P)的错义变异,即第2104位的氨基酸由亮氨酸变为脯氨酸,蛋白预测SIFT与Polyphen-2值分别为:0、0.997,预测该突变导致所编码的蛋白质发生构象改变从而影响正常功能。该家系患者均携带有此突变,遗传共分离。临床表型表现为脊椎椎管内神经纤维瘤,均无咖啡牛奶斑、虹膜Lisch结节、脊柱侧凸,无耳鸣、听力下降,无颅内压增高等。因此,我们认为NF1基因的c.T6311C(p.L2104P)错义突变是此NF1家系的遗传病因。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
神经纤维瘤病1型(neurofibromatosis 1,NF1,MIM:162200),又称Von Recklinghausen′s disease,是最常见常染色体显性遗传病之一,是由NF1基因缺陷导致神经嵴细胞发育异常继发的多脏器损害,除了经典特征如皮肤多发牛奶咖啡斑、腋窝或腹股沟雀斑、虹膜Lisch结节、周围神经多发性神经纤维瘤与骨损害等,还与恶性肿瘤、神经精神发育疾病(如认知功能障碍、焦虑、注意力缺陷多动障碍和自闭症谱系障碍)等有着密切的关系[1]。在NF1患者中曾发现视神经胶质瘤与性早熟[2],也曾发现罕见的并发溃疡性结肠炎[3],早期肺动脉高压也是NF1的严重并发症[4],可见该病临床表型谱宽广,临床较多异质性。在世界范围内,每3 000~3 500人中就有1例NF1患者[5],其致病的NF1基因被定位于染色体17q11.2,外显率高[3]。我们用第二代测序技术外显子捕获对以脊柱多发神经纤维瘤为特点的一个NF1家系进行遗传学分析,总结如下。
先证者(Ⅲ9)为男性,37岁,以"进展性左肢无力5年"为主诉于2016年9月到福建省立医院就诊,患者入院前5年无明显诱因出现左侧肢体乏力,行走跛行,无头晕头痛,无视物模糊、视物重影,无耳鸣、听力下降,无言语含糊、饮水呛咳,无肢体麻木疼痛,无意识不清、肢体抽搐、大小便失禁。症状进行性加重,行走愈加不稳,左上肢抬起费力,左下肢行走拖步,伴左侧肢体肌肉萎缩,无肌肉跳动。专科检查:脊椎生理曲度存在,无畸形。颈部无压痛、叩击痛,无棘突旁压痛,颈椎活动度可,前屈35°,后伸35°,左右侧屈45°,左右旋转30°。双侧Spurling试验阴性,双侧Eaton试验阴性,直腿抬高试验及加强试验阴性,余肢体及各关节活动度正常。神经系统体格检查:意识清楚,对答切题、言语清晰,高级皮质功能检查正常。脑神经检查正常。双上肢肌张力正常,双下肢肌张力增高,左上肢近端肌力Ⅳ级,远端肌力Ⅴ-级,左下肢肌力Ⅳ+级,右侧上下肢肌力Ⅴ级,左侧肢体肌肉萎缩,未见肌束颤动,站立时略向左侧偏斜,行走时左下肢略拖步,考虑左肢无力所致。右侧正常。双侧肢体深浅感觉及复合觉对称正常。双上肢腱反射(++),双膝腱反射(++++),跟腱反射(+++),双侧霍夫曼征阴性,左侧查多克征阳性,右侧查多克征阴性,双侧巴宾斯基征、高登征、奥本海姆征均阴性。脑膜刺激征阴性。经福建省立医院伦理委员会批准(伦审科研第K2015-007-01),家系成员签署知情同意书后提取DNA,对先证者NF1、NF2等单基因相关的43个致病基因的编码区及侧翼区进行目标区域捕获测序与生物信息分析。并对家系成员进行Sanger测序验证,NF1 (NM_000267)的exon41 (p.L2104P)的目标扩增长度为516 bp。引物为上游引物:5′-GCATAT-CTGTTTTATCATCAGGAGGTT-3′,下游引物:5′-GCAACTTGGTGTTAGAGCACAA-3′。用ABI 3730XL测序。
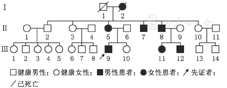
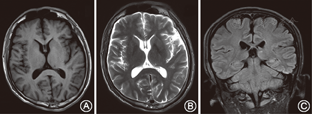
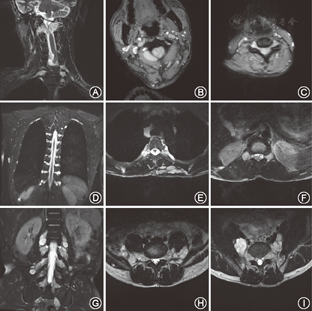
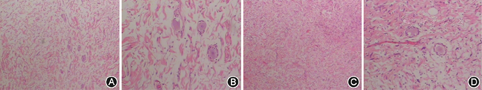
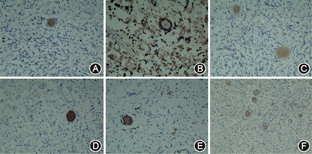
所调查的家系成员共15名,发病患者7例,均发现有肢体无力症状,MRI证实有脊椎多发的椎管内神经纤维瘤,而其他成员MRI均未发现椎管内神经纤维瘤。患者均无咖啡牛奶斑、虹膜Lisch结节、脊柱侧凸,无内脏肿块压迫出血症状,无耳鸣、听力下降,头晕,无持续性头痛、恶心、呕吐和视物不清等颅内压增高等临床表型(图1)。先证者(Ⅲ9)的MRI示左侧丘脑可见小斑片状长T1长T2信号,T2WI-FLAIR呈高信号;余双侧侧脑室旁白质内可见条状或类圆形长T1长T2信号,T2WI-FLAIR呈低信号;部分脑沟、池、室增宽。提示左侧丘脑高信号小病灶,轻度脑腔隙,轻度脑萎缩(图2)。双侧颈胸腰椎椎间孔及骶管扩大,双侧颈、胸、腰及骶神经根走行区见多发串珠状、丛状、囊状类圆形或梭形病灶,边界较清,长T1长T2信号,压脂T2像呈高信号。较大者位于颈1~2双侧椎间孔区,右侧较大,约2.5 cm×2.1 cm×1.8 cm,部分长入局部椎管内,明显压迫局部硬膜囊及脊髓。颈5~6右侧椎间孔区病灶部分长入椎管内右侧部,大小约1.3 cm×1.1 cm×1.8 cm,压迫局部硬膜囊及脊髓,向左侧移位。胸12至腰1左侧椎间孔区病灶部分长入局部椎管内,矢状位约0.9 cm×1.0 cm,局部脊髓圆锥稍受压移位。扫描区内胸腰背部皮质及部分肌肉间隙内见多发病灶,呈等T1等T2信号或长T1长T2信号,最大径约2.6 cm(图3)。颈1~2椎管外、颈5~6椎管内肿瘤切除术后病理:节细胞神经瘤(图4)。免疫组织化学:Ki67(1%+),S100(+++),神经元特异性烯醇化酶(节细胞+),Neu-N(节细胞+),Nf(节细胞+),SY(节细胞+)(图5)。
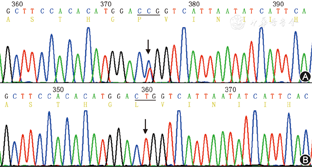
Sanger测序验证发现,该先证者携带了2个罕见变异,分别为NF1基因的外显子41上的c.T6311C杂合错义变异(NF1:p.L2104P het)和KIF1B基因c.C3649T杂合错义变异(KIF1B: p.P1217S het),其中NF1: p.L2104P是NF1的高度可疑致病突变,在NF1基因的外显子41上第2 104位的氨基酸由亮氨酸变为脯氨酸,预测该突变会导致所编码的蛋白质发生构象改变从而影响其正常功能。该突变未见文献报道,ESP6500、dbSNP和千人基因组数据库中均未见收录。并经Sanger验证(图6)。家系验证显示本家系7例发病患者中,除Ⅰ2已去世外,Ⅱ5、7、8及Ⅲ9、11、12均携带有p.L2104P错义突变;而Ⅱ2、3、10及Ⅲ5、7、8、10、13、14均未发现携带有此突变点。家系遗传分析说明了此突变点有遗传共分离现象。预测蛋白功能软件SIFT和Polyphen-2分数分别为0、0.997,提示此突变可能为致病性突变点。
NF临床表现主要有神经系统、皮肤与骨骼等发育生长异常,根据临床的表现,有NF1~7种类型,其中1~4型为常染色体显性遗传。NF1占NF发病者的90%,其突变的基因可导致患者于胚胎期2~4个月时神经外胚叶发育异常,男性多见。在Clinvar、HGMD数据库里,目前发现有2 000多个的致病突变。NF1基因由60个外显子组成,包括3个可变的剪接外显子,全长约350 kb,编码由2 818个氨基酸组成的NF1蛋白,于人胚胎发育中的间充质干细胞与神经嵴组织细胞以及全身组织广泛表达[6,7]。NF1蛋白有多个功能区,其中最重要的二个功能区域[7,8],一是高度保守的,含360个氨基酸的三磷酸鸟苷(GTP)酶超家族同源的与三磷酸鸟苷活性蛋白(GAP)相关的催化结构域(NF1-GRD),另一个则是富半胱氨酸/丝氨酸区域,有多个环腺苷酸依赖性蛋白激酶识别位点,位于GRD上游,起调控作用。目前,在NF1基因各外显子区域均发现有各种不同方式的突变,而在上述2个重要的功能域发现的突变众多,有报道在GRD区域的突变可引起GAP明显失活[9]。NF1基因跨度很长,贯穿整个基因都不断有新发现的突变点,没有明显的热点突变,从单个碱基突变到大片片段的缺失以及拷贝数变异[10],突变谱极广,这导致复杂的突变检出率不高,目前主要集中于点突变检出。NF1基因是人类突变率最高的基因之一,目前发现的突变方式有多个的错义/无义突变、剪切突变、片段缺失插入,还存在基因嵌合、假基因、二次打击再发突变等,大多NF1突变位置相对集中于外显子21-27与外显子11-17区域,超过70%的NF1基因突变为截断蛋白[9]。
常见的NF1相关肿瘤是良性的外周神经鞘瘤或神经纤维瘤,小部分NF1患者的丛状神经纤维瘤可进展到恶性周围神经鞘瘤,染色体结构不稳定是导致恶性变的主要因素。而周围神经系统施万细胞缺陷是神经纤维瘤发展的基础,NF1患者也易患脊髓肿瘤、嗜铬细胞瘤、星形细胞瘤、青少年髓样单核细胞白血病、Watson综合征和胃肠道间质瘤[11]。NF1蛋白作为Ras信号转导通路的负调控因子发挥作用,而Ras-GAP功能的失活与临床表型明显相关[12]。GRD是目前研究神经纤维瘤病最好的切入领域,其作为负调控因子,可将活性RAS-GTP转化为无活性RAS-鸟苷二磷酸(RAS-GDP),从而抑制下游RAS信号。在一些错义突变的NF1患者中,其突变并没有影响蛋白质构象与水平,而是选择性地减弱了RAS-GTP酶活性[13]。
本家系的NF1患者携带了杂合突变p.L2104P,该变异在千人基因组、ESP6500数据库和ExAC数据库均无人群频率报道,可明确其为罕见变异;SIFT和Polyphen-2软件交叉预测均提示该变异所致的氨基酸改变会对蛋白产生影响,该氨基酸在脊椎动物中高度保守。p.L2104P曾在一个阿拉伯人的NF1家系中被发现,该家系中的父亲跟儿子均为杂合携带者,但仅儿子为NF1患者,患病年龄不详,2例均有钙损伤或钙斑点,均无咖啡牛奶斑、虹膜Lisch结节、丛状神经纤维瘤和脊柱侧凸等临床表型[14]。同一位点的变异p.L2104R曾在NF1患者中也被发现,但具体临床表型不详[15]。此外,尚有同一位点的变异p.L2104V被ClinVar数据库收录,但尚未上传NF1的具体临床表型。因此,结合目前现有证据及家系分析,该变异可判为NF1的可疑致病变异。
此家系先证者还携带了KIF1B基因c.C3649T(p.P1217S)杂合错义变异,该变异千人基因组及ExAC数据库均人群频率较低,ESP6500数据库的人群频率为0,也可明确其为罕见变异;SIFT和Polyphen-2软件交叉预测提示该变异所致的氨基酸改变对蛋白产生影响的结果不一致,该氨基酸在哺乳动物中高度保守。此变异被ClinVar收录,上传者将其判定为神经母细胞瘤的"风险因子(risk factor)";也有1例散发的神经母细胞瘤患者携带该变异,而270名对照人群未携带该变异[16]。尽管神经母细胞瘤在儿童的发病率高,但也有成人发病的,本家系未发现有此病患者,但仍需密切随诊。仅从本研究结果来看,p.L2104R似乎在这个汉族家系中只表现为脊椎多发的椎管内神经纤维瘤。
无
None declared

























