
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
糖尿病出现血糖稳态失衡的主要病因之一是伴或不伴胰岛素抵抗的胰岛功能障碍。在2型糖尿病(T2DM)早期,β细胞通过体积增大、数量增多、分泌能力增强以应对胰岛素抵抗带来的胰岛素需求。然而长期超负荷工作最终会导致β细胞功能衰竭与高血糖的发生。研究发现,在此过程中,不仅胰岛内部各细胞间存在复杂的信号交流,外周组织脏器如脂肪、肝脏、骨骼肌、肠道等所释放的激素、调控因子、代谢产物均参与了与胰岛的通讯(图1)。这些研究结果也为促进β细胞新生、恢复胰岛功能提供新的治疗思路与靶点。现就胰岛与机体多器官间的通讯作一概述。
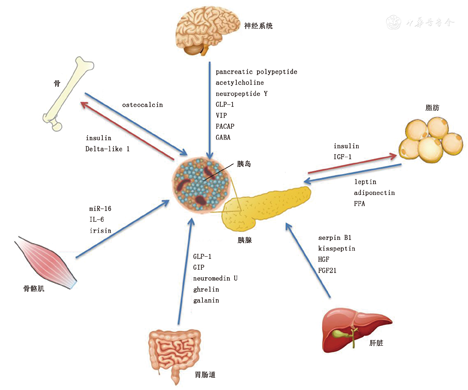
注:osteocalcin:骨钙蛋白;insulin:胰岛素;Delta-like 1:δ样1蛋白;IL-6:白细胞介素6;irisin:鸢尾素;miR-16:微小核糖核酸16;pancreatic polypeptide:胰多肽;acetylcholine:乙酰胆碱;neuropeptide Y:神经肽Y;GLP-1:胰高糖素样肽-1;VIP:血管活性肠肽;PACAP:垂体腺苷酸环化酶激活肽;GABA:γ-氨基丁酸;GIP:葡萄糖依赖性促胰岛素多肽;neuromedin U:神经调节肽U;ghrelin:胃饥饿素;galanin:肝丙肽;IGF-1:胰岛素样生长因子-1;leptin:瘦素;adiponectin:脂联素;FFA:游离脂肪酸;serpin B1:中性粒细胞弹性蛋白酶抑制剂B1;HGF:肝细胞生长因子;FGF21:成纤维生长因子21;
脂肪细胞参与内分泌代谢调节的作用已逐渐被阐明。脂肪细胞出现脂质沉积、肥大增生的同时,伴随脂联素分泌的减少与瘦素、促炎因子分泌的增加,这些体液因素的改变调控β细胞的功能,并参与胰岛素抵抗的发生发展。
脂肪细胞可释放胞内过多的游离脂肪酸进入血循环,通过内质网应激和活性氧簇对β细胞产生脂毒性,激活多种促炎细胞因子的表达。瘦素除作用于下丘脑调节人类食欲外,还可独立作用于β细胞抑制胰岛素的产生与分泌。然而,瘦素对于胰岛功能的影响具有两面性。在正常生理代谢状态下,瘦素抑制β细胞增生与胰岛素分泌;而高脂诱导肥胖状态下,血浆高水平瘦素可改善糖耐量、促进胰岛素分泌,参与β细胞的适应性改变[1]。脂联素作为脂肪细胞来源的另一种激素,可保护β细胞免受脂毒性损伤,并通过内源性及外源性信号通路抑制β细胞凋亡,促进胰岛新生。在1型糖尿病(T1DM)中,脂联素还可下调树突状细胞表面共刺激分子的表达从而减轻自身免疫反应,延缓β细胞损伤。
近年来关于脂肪、脑、骨与β细胞的研究进一步证实了胰岛多脏器间的协同作用。瘦素可通过下丘脑增加交感兴奋性,负性调控骨钙蛋白的生物活性,最终影响β细胞功能;而脂联素可降低交感兴奋性,对β细胞产生相反于瘦素的作用。多脏器间的相互交流与反馈,多角度多方面维系着胰岛功能的稳态。
此外,胰岛对脂肪组织执行正常生理功能也不可或缺。白色或棕色脂肪中胰岛素或胰岛素生长因子-1受体缺乏,可引起脂解与脂肪细胞凋亡,并最终出现糖耐量受损、胰岛素抵抗、胰岛增生与脂肪肝等[2],由此可见脂肪与胰岛的信息交流是双向的且十分精密。
肝脏是一个很理想的可与胰岛进行通讯的器官,首先肝脏与胰岛有共同的胚胎起源;其次,解剖结构上两者共属于门静脉系统,代谢产物、激素等体液因子的交流更加频繁;再者,肝脏作为能量储存、糖脂代谢的主要器官,与胰岛素抵抗、T2DM的发生密切相关。利用联体共生和胰岛移植技术,许多研究均报道了肝脏来源的某些因子可直接影响胰岛功能和机体代谢,近年来的后续研究中发现该种因子可能是肝脏中性粒细胞弹性蛋白酶抑制剂(serpinB1),并证实其可促进鼠、斑马鱼和人类β细胞增殖[3]。其他肝脏来源因子,如肝细胞生长因子可作用于β细胞表面的c-Met受体,从而改变细胞增殖能力[4],成纤维细胞生长因子-21可促进糖尿病小鼠胰岛素分泌,抑制胰高糖素水平,阻止胰岛炎的进展[5]。
除肝脏与β细胞相互作用外,还存在"肝脏-α细胞轴(hepatic-α cell axis)"[6],胰高糖素作用于肝脏促进氨基酸合成利用,而肝细胞产生的氨基酸尤其是左旋谷氨酰胺,可通过哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)通路及叉头框P(forkhead box P)转录因子促进α细胞增殖。
由KISS-1基因编码的kisspeptin也参与了肝脏胰岛间通讯,kisspeptin首先是在非侵袭性黑色素瘤细胞中发现的,而其参与糖代谢的相关作用近几年来引起研究者们较大的兴趣。α细胞分泌的胰高糖素可通过环腺苷酸(cyclic adenosine monophosphate,cAMP)-蛋白激酶A(protein kinase A,PKA)-cAMP效应元件结合蛋白(cAMP-response element binding protein,CREB)通路激活肝脏kisspeptin的表达,其作用于β细胞表面的kisspeptin受体后可抑制胰岛素分泌[7]。kisspeptin将α细胞-肝脏-β细胞整个内分泌代谢回路相连接,为肝脏胰岛多组分间的相互作用提供有力证据。
胰岛的神经网络错综复杂,α、β、δ等细胞周围分布着丰富的交感与副交感神经共同调控血糖稳态。值得注意的是,小鼠和人类胰岛内神经的分布模式存在显著差异,小鼠胰岛核心以副交感神经为主,而交感神经主要分布在胰岛外周;相比之下,人类胰岛交感神经随血管分布延伸于整个胰岛,而副交感神经却较少贯穿,人类α细胞可替代分泌胆碱能神经递质作用于周围的β细胞。神经与其他体液因素最大的不同之处,在于对靶器官调节速度之迅速。由于细胞系或原代胰岛上模拟神经递质调节作用难度较大,此类研究急需新型模拟内环境的胰岛立体体外培养技术。
β细胞外排胰岛素与神经组织分泌突触递质上有许多相似之处,同为神经内分泌组织,其腺苷三磷酸(adenosine triphosphate,ATP)敏感性钾通道、钙离子内流等都有共通点。所有在中枢神经系统发现的神经递质在胰岛均有表达,就像在不同神经元一样,这些神经递质聚集积累在不同细胞中,例如α细胞中的谷氨酸、乙酰胆碱,β细胞中的γ-氨基丁酸、ATP、5-羟色胺、多巴胺等,δ细胞中的生长抑素,ε细胞分泌的胃饥饿素(ghrelin)。除神经递质外,多种神经肽也参与了胰岛调控,包含肠促胰素家族中的胰高糖素样肽-1(GLP-1)、垂体腺苷酸环化酶激活肽(pituitary adenylate cyclase activating peptide)、血管活性肠肽(vasoactine intrestinal peptide)等,以及胰多肽家族中的神经肽Y、胰多肽等。
大脑参与维持糖稳态的作用早在19世纪就已被阐明,近年来有学者提出了以脑为中心的葡萄糖调节系统(brain-centred glucoregulatory system),其可通过胰岛素依赖性和非依赖性机制与胰岛共同调节糖稳态[8]。大脑同胰岛一样,也表达葡萄糖转运蛋白2,后者被敲除后胰岛素分泌能力受损,糖耐量下降,β细胞的体积与增殖能力也同时降低。近年来有研究利用伪狂犬病病毒追溯胰岛内的神经来源,提示胰岛神经主要来源于下丘脑,下丘脑的不同核团与脑区可参与调控胰岛素、胰高糖素的分泌[9]。此外,下丘脑的脂毒性损伤也参与了中枢胰岛素抵抗,棕榈酸可促进下丘脑神经酰胺的合成,并由此诱发胰岛素抵抗,而神经酰胺抑制剂的应用可逆转棕榈酸所带来的糖耐量受损,并增加β细胞数量[10]。然而除下丘脑外,其他脑区如大脑皮层、中脑、脑桥、延髓等部分与胰岛间的信息交流仍有待研究。
早年研究就发现肌肉特异性PGC1α敲除鼠存在糖耐量受损,其中白细胞介素6(interleukin 6,IL-6)可能是参与β细胞胰岛素分泌能力下降的关键分子。但是,关于肌肉组织IL-6对胰岛的影响现仍存在争议,有研究表明IL-6敲除的高脂喂养肥胖鼠存在胰岛素分泌障碍,提示IL-6有助于胰岛素抵抗状态下β细胞代偿分泌。此外,骨骼肌分泌的IL-6可刺激肠道L细胞分泌GLP-1,同时还可通过转录后修饰使α细胞胰高糖素原更多地加工合成GLP-1而非胰高糖素[11]。此外,Irisin作为骨骼肌分泌的一种细胞因子,除可将白色脂肪转化为棕色脂肪外,体内及体外研究均证实可通过PKA依赖性途径促进胰岛素分泌及β细胞增殖,阻止饱和脂肪酸等脂毒性所致的胰岛β细胞凋亡[12]。
由外泌体介导的细胞间通讯正逐渐被人关注,这些微结构中的微小核糖核酸(microRNA,miR)、蛋白质不仅可作为生物标记物,同时可向靶细胞传递重要的生物信息。MIN6B1细胞系及原代胰岛经高脂饮食鼠骨骼肌外泌体处理后,β细胞出现增殖,其中起作用的可能是miR-16及其下游Patched1相关通路[13]。
骨作为一种内分泌组织影响机体代谢已逐渐被人所认可。骨钙蛋白(osteocalcin)是一种成骨细胞特异性合成分泌的蛋白,其可通过谷氨酸残基的羟基化,结合羟磷灰石、钙沉积在骨的细胞外基质。而去羟基化的骨钙蛋白与羟磷灰石的结合力显著减弱,更易释放进入血循环,从而作为一种内分泌因子影响其他组织功能代谢。
骨钙蛋白可促进β细胞增殖与胰岛素分泌能力,并提高胰岛素敏感性,其对于β细胞的作用主要是通过β细胞表面的Gprc6a受体完成的,而骨睾丸蛋白酪氨酸磷酸酶(osteo-testicular protein tyrosine phosphatase,OST-PTP)可通过γ-羟基化负性调控骨钙蛋白对胰岛的作用。胰岛对于成骨细胞的影响也不容忽视,β细胞分泌胰岛素可作用于成骨细胞表面胰岛素受体,抑制Twist2蛋白的表达,从而促进骨钙蛋白的分泌,而OST-PTP可通过成骨细胞胰岛素受体的去磷酸化中断胰岛素对骨钙蛋白合成的作用。此外,δ样1(delta-like 1,DLK-1)也参与胰岛与骨钙蛋白之前的前馈调控,骨钙蛋白刺激β细胞DLK-1的表达,DLK-1与胰岛素一起共分泌,DLK-1也在成骨细胞中抑制胰岛素受体的信号转导[14]。
GLP-1与葡萄糖依赖性促胰岛素多肽(glucose-dependent insulinotropic polypeptide)是肠促胰素(incretin)中最广为人知的两种激素,分别是由肠道L与K细胞分泌的,并动态地根据体内糖脂代谢水平调节胰岛素的分泌。GLP-1在刺激β细胞胰岛素分泌的同时,还可作用于α细胞GLP-1受体抑制胰高糖素的分泌,最终被内皮细胞二肽基肽酶Ⅳ降解。值得一提的是,β细胞特异性GLP-1受体敲除鼠并不会阻断GLP-1促胰岛素分泌的作用,这提示GLP-1可能是作用于肠道或门静脉系统的传入神经GLP-1受体,最终将胰岛素分泌信号传递至β细胞,而并非直接通过β细胞表面的GLP-1受体。除肠促胰岛素外,肠降胰岛素(decretin)也是一类由肠道产生抑制胰岛素分泌的激素,包括神经调节肽U(neuromedin U)、ghrelin、甘丙肽(galanin)等。胰岛单细胞测序结果表明多种食欲调控相关激素如ghrelin等的受体选择性的高表达于胰岛δ细胞,提示进食等相关因素可能通过影响δ细胞生长抑素的分泌,从而调节α细胞与β细胞间的平衡。
肠道微生物的改变与肥胖、T2DM等代谢类疾病密切相关,肠道菌群移植可改善胰岛素敏感性。远端肠管内微生物对食物残渣的发酵所产生的短链脂肪酸,是目前被认为主要影响代谢的因素。β细胞表达游离脂肪酸受体2(也称为G蛋白偶联受体43),缺乏该受体的敲除鼠在高脂喂养后出现显著的糖耐量下降、胰岛素分泌能力受损、β细胞数量减少[15]。此外,肠道菌群可影响肠黏膜的通透性,通过分子模拟等方式参与固有免疫与适应性免疫,在T1DM的发生发展中起重要作用。
尽管目前糖尿病治疗新药层出不穷,然而针对阻止T2DM患者β细胞衰竭进程的药物屈指可数。当发生胰岛功能障碍、胰岛素抵抗时,来自各个脏器的多种信号相互整合、共同影响胰岛,作用于能量代谢、内质网应激、氧化应激、促炎级联反应等,产生的恶性循环、前馈反应促成胰岛素分泌能力受损、β细胞凋亡甚至去分化。在胰岛多脏器通讯的研究中,我们还有更大的空间等待探索:(1)胰岛内部α、β、δ、ε、PP细胞共同协作维持糖稳态,而目前大多研究仅局限于与β细胞间的通讯,其他细胞与多脏器的交流有待发现;(2)在糖尿病发病的不同时期,哪些器官的哪种物质在多胰岛多脏器交流中起主导作用,而哪些作用处于次要地位;(3)如何将现阶段在动物水平观察的相关结果转化并应用于人类糖尿病的病理生理研究;(4)在未来糖尿病分型中,是否可以分以β细胞自主损伤为主与β细胞被动功能下降为主的多种亚型。以上这些问题都是亟待我们研究的。

























