
Dent病是一种罕见的X连锁遗传性肾小管疾病,其临床特征为低分子质量蛋白尿(LMWP)、高钙尿、肾钙化和/或肾石症,可有其他近端肾小管功能异常和肾功能异常。现已知Dent病Ⅰ型致病基因为编码氯离子通道-5的CLCN5基因,Dent病Ⅱ型致病基因为编码磷脂酰肌醇5-磷酸酶的OCRL1基因。其肾脏病理类型常见局灶节段性硬化(FSGS)、系膜增生性肾小球肾炎(MsPGN)、微小病变(MCD),30~40年可进展至慢性肾衰竭。作为Dent病关键点的LMWP,在儿童常呈肾病水平蛋白尿,但血清清蛋白正常,在临床上应注意与肾病综合征鉴别。当男童有LMWP伴高钙尿或肾钙化时应送检CLCN5和OCRL1基因检测,以避免误诊和漏诊Dent病。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
现代肾脏病学发展至今,在对肾小球疾病深入研究的同时,对于肾小管-间质相关疾病也日益关注;随着分子生物学研究及基因检测技术的日臻完善,越来越多的遗传性肾脏病在临床得以诊断,其中也包括遗传性肾小管疾病。在此,我们将儿科较为常见的一组遗传性肾小管疾病,结合临床诊治体会予以介绍,旨在提高临床医师对于该类疾病的认识,减少漏诊与误诊。
随着对肾小管疾病的日渐重视及基因诊断技术的快速发展和临床应用,越来越多的遗传性肾小管疾病已为临床认知和确诊,其中Dent病则是儿科临床中相对常见的遗传性肾小管疾病之一。作为X连锁隐性遗传性肾小管疾病,Dent病以低分子质量蛋白尿(low mole-cular weight proteinuria,LMWP)、高钙尿症、高磷酸尿乃至Fanconi综合征等近端肾小管功能异常为主要表现。自1964年Dent等首先报道至今,全球已报道超过300个家系。我国自2008年北京大学第一医院儿科报道首例基因确诊儿童Dent病以来[1],多家医院陆续有病例报道,迄今已报道病例54例(表1)。有关Dent病的病例报道及综述不断见诸各种专业杂志,反映出目前国内儿科对于Dent病已有相当的重视和认知。作为一种罕见的X连锁隐性遗传性肾小管疾病,本症可在全儿科阶段发现,但常因认识不足、检查不到位及表现不典型而漏诊或误诊。

我国已报道儿童Dent病基本情况
The basic situation of children with Dent disease had been reported in China
我国已报道儿童Dent病基本情况
The basic situation of children with Dent disease had been reported in China
| 发表单位 | 年份 | Dent类型 | 例数 | NRP例数(例) | 肾活检例数(例) | 肾脏病理 | 其他(例) |
|---|---|---|---|---|---|---|---|
| 北京大学第一医院 | 2008/2010 | Ⅰ | 6 | 4 | 1 | TIN | |
| 上海复旦大学附属儿科医院 | 2014 | Ⅱ | 1 | 1 | Miner | CPK高1例 | |
| 上海复旦大学附属儿科医院 | 2015 | Ⅰ | 5 | 2 | |||
| 北京协和医院 | 2015 | Ⅰ | 4 | 4 | 1 | FSGS | Fanconi综合征3例 |
| 北京军区总医院附属八一儿童医院 | 2015 | Ⅰ | 15 | 5 | 10 | FSGS-1、MsPGN-2、Miner-7 | |
| 南京市儿童医院 | 2016 | Ⅰ | 1 | 1 | |||
| 首都儿科研究所附属儿童医院 | 2017 | Ⅰ | 3 | 2 | |||
| 上海交通大学附属儿童医院 | 2017 | Ⅰ | 6 | 3 | 1 | FSGS | |
| 北京大学第一医院 | 2017 | Ⅰ | 9 | 9 | 5 | FSGS-2、MCD-2、MsPGN-1 | |
| 北京大学第一医院 | 2017 | Ⅱ | 1 | 1 | 白内障+智力低下1例 | ||
| 北京市儿童医院 | 2018 | Ⅰ | 3 | 2 | 2 | MsPGN-1、FSGS-1 |
注:NRP:肾病水平蛋白尿;CPK:磷酸肌酸激酶;TIN:肾小管间质肾炎;Miner:轻微病变;FSGS:局灶节段性硬化;MsPGN:系膜增生性肾小球肾炎;MCD:微小病变 NRP:nephropathy-range proteinuria;CPK:creatine phosphate kinase;TIN:tubulointerstitial nephritis;Miner:Miner change;FSGS:focal segmental sclerosis;MsPGN:mesangial proliferative glomerulonephritis;MCD:minimal change
在此,笔者结合国内外文献及北京大学第一医院儿科在儿童Dent病临床实践中的经验体会,从临床角度出发,结合致病基因与发病机制,对Dent病在儿科的临床诊断及治疗预后等系列临床相关问题作一介绍,旨在供临床医师在日常工作中参考,以减少漏诊和误诊。
(1)CLCN5基因:1996年由Lloyd等[2]报道了位于Xp11.22-p11.23的CLCN5基因是Dent病的致病基因,其编码的蛋白ClC-5是氯离子通道蛋白。迄今已报道可导致Dent病的CLCN5基因突变共192种,其突变类型以错义突变和移码突变为主,占比分别为36.5%和28.0%;其他类型包括无义突变(17.0%)、剪切突变(11.0%)和大片段缺失(4.0%)[3]。CLCN5基因突变可导致ClC-5蛋白截断或缺失,进而导致其功能丧失,从而影响到近端肾小管上皮细胞氯离子电导的损耗及逆向转运蛋白功能缺陷,损伤囊泡的酸化功能,造成近端肾小管细胞胞吞作用障碍[4],引发一系列Dent病的临床异常:①原尿中清蛋白和低相对分子质量蛋白的重吸收功能障碍导致LMWP。②通过影响集合管闰细胞(intercalated cells)的尿液酸化功能,改变尿液pH值,促进肾结石形成。③通过影响Megalin/Cubilin受体依赖形式的胞吞作用,使得原尿中的VitD结合蛋白、25(OH)-D3和甲状旁腺激素(PTH)在近端肾小管的重吸收障碍;进而通过增加的远端肾小管腔内尿中PTH水平,高PTH水平一方面激活顶膜侧的PTH受体,PTH受体可激活刷状缘的钠-磷共转运蛋白NaPi-Ila,致使近端肾小管磷的重吸收减少,产生高磷酸盐尿、低磷血症表现[5];另一方面通过影响PTH对细胞膜的极化作用刺激线粒体1α羟化酶的转录,促进1,25(OH)2-D生成增加,引发吸收型高钙尿症。高尿磷酸盐、高尿钙和尿pH值改变共同影响Dent病肾钙化和肾结石的发生。(2)OCRL1基因:2005年Hoopes等[6]报道了与Dent病相关的第2个致病基因。该基因位于染色体Xq25,有24个外显子(exon),该基因在人体组织中广泛存在,尤其在眼睛、肾脏和大脑,其编码蛋白为磷脂酰肌醇4,5-二磷酸5-磷酸酶,该酶可以水解磷脂酰肌醇4,5-二磷酸(PIP2)。PIP2属于跨膜蛋白,在肾脏主要分布在肾小球、近端肾小管和集合管,其中在近端肾小管上皮细胞中主要分布于高尔基复合体、溶酶体和早期内涵体内,具有跨膜转运调节作用,参与胞吞作用,在Dent病时可导致LMWP;PIP2通过肠上皮Ca2+通道的瞬时受体电位阳离子通道蛋白6(TRPV6)途径影响肠道对Ca2+吸收,在Dent病时产生高钙尿症。OCRL1基因也是已知眼-脑-肾综合征(即Lowe综合征)的致病基因,故由该基因突变引起的Dent病 Ⅱ 型亦可出现Lowe综合征样的肾外表现。
目前Dent病患者发现的OCRL1基因突变约140余种,包括插入、缺失、剪切和错义突变,其中错义突变主要分布于exon 4~15,而剪切突变主要位于exon 1~7或内含子7上,导致读码框移位和终止密码提早出现[7,8,9]。
现依据致病基因的不同将Dent病分别命名为:Dent病 Ⅰ 型(OMIM# 300009,由CLCN5基因突变所导致,约占已知Dent病的60%,是Dent病的主要类型)和Dent病 Ⅱ 型(OMIM#300555,由OCRL1基因突变所导致,约占已知Dent病的15%,迄今全球报道病例不足50例)[6]。
目前仍有25%的Dent病未检测出CLCN5和OCRL1基因突变,其确切的致病基因不详[10]。与ClC-5同属于哺乳类ClC家族中的第二亚族(酸化功能调节型ClC通道蛋白)的ClC-4亦是2Cl-/H+逆向转运体,该蛋白的基因位于Xq22.3,当第224位谷氨酸突变后,将失去2Cl-/H+逆向转运功能,很有可能与Dent病相关[11]。
国内外学者均观察到Dent病的基因型与临床表型无明确相关性,本症具有明显的遗传异质性。北京大学第一医院观察到一家系同胞兄弟检出相同的CLCN5致病突变,临床除LMWP和高钙尿外,在伴肾钙化、佝偻病、氨基酸尿、电解质紊乱等多方面表现明显不同[12]。
目前在临床上已知Lowe综合征和Dent病 Ⅱ 型均由OCRL1基因突变所致,同一致病基因导致2个不同疾病的原因何在?现有解释如下:主要是2个病症的基因突变位点差异所致。已有文献资料显示:导致Dent病Ⅱ型的基因突变位点主要分布在OCRL1基因exon 1~15区域,其中错义突变位点分布在exon 4~15,剪切突变分布在exon 1~7以及内含子7[9]。而导致Lowe综合征的OCRL1基因突变的位点主要分布在exon 15~23区域[13,14]。或许正是因为这种突变位点的差异,使得Dent病 Ⅱ 型表现出类似Lowe综合征样的临床表现,而在高钙尿、肾石症方面强于Lowe综合征,在代谢性酸中毒、糖尿、氨基酸尿、肾功能减退及智力低下、眼症方面逊于Lowe综合征。所以,笔者认为未来以致病基因命名OCRL1相关疾病,分为Lowe亚型和Dent亚型,而Dent病为CLCN5基因突变所致更为适合临床。
尚见有CLCN5和OCRL1 2个致病基因突变导致儿童不典型Dent病的报道[15]。
自1964年Dent等报道了2例患儿表现为肾小管性蛋白尿、氨基酸尿、高钙尿等肾小管功能障碍后,陆续有相似临床表现的综合征报道,如X连锁隐性肾石病、X连锁隐性遗传性低磷酸盐血症性佝偻病、X连锁高钙尿性肾结石病和日本儿童特发性LMWP等相继出现,这些综合征被认为是同一疾病的不同表现类型。Wrong等[16]于1994年报道该类患者还可发生肾石病、肾功能不全,并将这一系列综合征统称为Dent病。Dent病主要临床表现为LMWP、高钙尿,部分可有肾钙化或肾结石及各种近端肾小管功能异常表现,可出现进行性肾功能异常,最终发展至终末期肾病(ESRD)。
目前国内外通常采用以下临床诊断标准:(1)LMWP,即尿蛋白电泳提示低分子蛋白>50%,或尿α1微球蛋白、β2微球蛋白和/或视黄醛结合蛋白(RBP)升高至少高于正常上限5倍,且以低分子蛋白为主;(2)高钙尿症,即24 h尿钙>4 mg/kg(>1 mmol/kg)或随机尿钙/尿肌酐比>0.25 mg/mg;(3)至少有下列情况之一:肾钙化、肾石症、血尿、低磷血症或肾功能不全[5,16,17,18]。满足条件(1)(2)(3)者可临床确诊Dent病。
日本学者提出具有上述诊断条件(1)或(2)加(3)即可临床诊断[19]。尚见仅有条件(1)或(2)加CLCN5或OCRL1基因突变诊断Dent病(Ⅰ、Ⅱ 型)的报道[20,21,22,23,24]。对此笔者认为:从Dent病认识的发展过程看,这些与钙磷代谢异常的临床综合征(X连锁隐性肾石病、X连锁隐性遗传性低磷酸盐血症性佝偻病、X连锁高钙尿性肾结石病)均是经CLCN5基因检测纳入Dent病的;而CLCN5和OCRL1基因突变引发的基本病生理改变均为LMWP,高钙尿的机制尚不清晰,因此,仅有条件(2)加(3)在诊断Dent病时应该有基因检测的支持才能确诊。条件(1)加(3)的情况亦同样。目前国内所报道Dent病的诊断标准均为严格满足条件(1)(2)(3),并检出CLCN5或OCRL1致病性基因突变。
当检测到OCRL1基因突变时,应注意合并肾外表现的检查,发现轻度智力障碍、白内障及血磷酸肌酸激酶(CPK)、乳酸脱氢酶(LDH)升高等表现时考虑Dent病 Ⅱ 型,但注意与Lowe综合征鉴别(表2)。
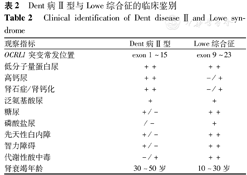
Dent病 Ⅱ 型与Lowe综合征的临床鉴别
Clinical identification of Dent disease Ⅱ and Lowe syndrome
Dent病 Ⅱ 型与Lowe综合征的临床鉴别
Clinical identification of Dent disease Ⅱ and Lowe syndrome
| 观察指标 | Dent病 Ⅱ 型 | Lowe综合征 |
|---|---|---|
| OCRL1突变常发位置 | exon 1~15 | exon 9~23 |
| 低分子量蛋白尿 | ++ | ++ |
| 高钙尿 | ++ | -/+ |
| 肾石症/肾钙化 | ++ | -/+ |
| 泛氨基酸尿 | + | + |
| 糖尿 | +/- | ++ |
| 磷酸盐尿 | /- | + |
| 先天性白内障 | +/- | ++ |
| 智力障碍 | +/- | ++ |
| 代谢性酸中毒 | -/+ | ++ |
| 肾衰竭年龄 | 30~50岁 | 10~30岁 |
Dent病可能是临床Fanconi综合征、局灶节段性硬化(FSGS)的病因之一,确切说是Fanconi综合征、FSGS可以是Dent病的临床-病理表现形式(表1)。北京大学第一医院曾见Bartter综合征样的Dent病 Ⅰ 型1例[25],与Besbas等[26]的报道类似。因此,临床上对于患有FSGS、Fanconi综合征、Bartter综合征(氯离子通道CLC-κb异常所致)乃至肾结石/肾钙化、低磷抗D佝偻病的男童,应注意寻找相关的临床证据,如送检基因检测注意包括CLCN5和OCRL1基因,以免漏诊Dent病。
有学者提出,因为多数Dent病预后较好,且基因型与临床表现间没有必然的相关性,故不推荐对Dent病进行产前诊断和基因检测[27]。笔者认为,虽然Dent病的基因型与临床表型无相关性,且儿童期少有肾功能减退,但致病性基因突变的遗传会导致子代男性罹患本症可能,以及成人阶段后存在肾衰竭/ESRD的风险,故除非患儿是自发基因突变或检测鉴定为女性胎儿,再次怀孕时产前诊断有其必要性。
总之,即使临床具备Dent病诊断(1)(2)(3)条标准,CLCN5或OCRL1基因致病突变的检测对于Dent病的诊断与分型仍起着决定性作用。
蛋白尿是Dent病核心的临床表现,发生率近乎100%,呈现为LMWP,即使定量为肾病水平蛋白尿(nephrotic-range proteinuria,NRP)时,其蛋白尿性质仍为肾小管性蛋白尿,低相对分子质量蛋白占比>50%,临床也基本不引起血浆清蛋白减低及水肿、高脂血症表现,可与肾病综合征(NS)区别。通常认为LMWP的定量<1 g/24 h,但儿童Dent病中NRP较为常见,在我国儿科已报道的54例Dent病中32例有NRP,约占59.3%。目前就Dent病伴NRP大致有如下的解释:(1)从Dent病的肾活检病理结果看均存在肾小球病变,以FSGS和微小病变/轻微病变(MCD/Miner change)为主[28](表1)。这些肾小球病变可能参与肾小球性尿蛋白成分的漏出,造成总尿蛋白定量的增加。(2)国外学者通过WT1与ClC-5共定位技术观察到在人类肾小球(足细胞)存在ClC-5在mRNA和蛋白质水平的表达,且在肾小球性蛋白尿患者肾小球足突融合处存在ClC-5的高表达[29,30]。鉴于足细胞在FSGS、MCD的蛋白尿发生中发挥的作用,提示ClC-5蛋白功能的异常很可能通过对足细胞的影响参与了肾小球性蛋白尿的产生。北京大学第一医院在Dent病肾病理为FSGS和Miner change的电镜下均见到节段性足突融合表现[28,31]。(3)国内外有学者提出Dent病NRP是病情发展的观点,即蛋白尿程度不同考虑与肾脏受累程度不同有关,随着病程发展,均可在肾小管疾病的基础上继发性出现肾小球损害而出现大量蛋白尿[21,32]。Valina等[33]对Dent病患儿重复肾活检显示2年后肾病理从Miner change转为FSGS的报道也支持这一观点。(4)罕见伴发NS状态。北京大学第一医院曾报道1例Dent病 Ⅱ 型伴发激素敏感型原发性NS。该患儿以NS发病,规范足量激素治疗4周后尿蛋白++,呈现"激素耐药",联合多种免疫抑制剂治疗后,水肿消退,血清蛋白及胆固醇均已正常,但24 h尿蛋白定量仍波动在0.41~0.53 g。院内检查为小管性蛋白尿(尿蛋白电泳示小分子蛋白64.4%)伴泛氨基酸尿、高钙尿,肾脏病理为Miner change,电镜下见节段性足突融合,送检基因检测证实,OCRL基因c.2435T>C(p.L812P)纯合突变,最终结合白内障及Gesell评分异常确诊Dent病 Ⅱ 型伴原发性NS(激素敏感型)[31]。
LMWP是临床考虑Dent病的切入点之一,日本3岁以上儿童国家尿筛查计划中不乏早期检出Dent病的病例(12例,4个家系)[34]。而目前儿科医师(包括儿科肾脏病医师)临床上对蛋白尿的关注常常是"习惯性"视同肾小球病的结果,重尿蛋白定量,轻尿蛋白性质,LMWP漏诊现象易见。在北京大学第一医院发表的Dent病6例临床诊治分析看到,全部患儿在院外均忽略了对大量蛋白尿的定性检测,诊断"激素耐药型NS"而接受相关治疗,从而导致Dent病的漏诊与误诊[35]。因此,有必要再度强调:对于儿童蛋白尿在注意定量的同时,一定要关注蛋白尿性质,切不要忽略肾小管性蛋白尿,尤其是按常规治疗后尿蛋白改善不满意时。鉴于目前国内许多医院尚未开展尿蛋白电泳检测,而肾早期损伤指标检测较为普及的现状,本研究提出注意尿肾早期损伤指标中α1微球蛋白、β2微球蛋白及视黄醛结合蛋白等小分子质量蛋白与微量清蛋白的比值(>1)有助于及早发现LMWP。
Dent病作为X连锁隐性遗传病,主要累及男性,但目前也见少数有关女性Dent病的报道[36,37]。所谓女性Dent病患者是指有Dent病致病基因检出,同时具有一定Dent病临床表现的女性携带者,通常仅表现为部分症状,且其临床症状较男性Dent病轻或不典型,约70%的女性携带者仅有轻度LMWP,50%存在高钙尿;少见严重的肾脏钙沉着症、肾石症及ESRD的病例报道[38]。
有关女性Dent病发生的机制尚不完全清楚。Minamikawa等[37]通过超深定向RNA测序方法(ultra-deep targeted RNA sequencing)已证实近50%的女性Dent病是由X染色体失活偏移所致。另有推测可能与女性携带者非致病基因X染色体缺失(45,X)或双等位致病基因携带(即来自母亲携带者和患病的父亲)有关,至今尚未得到证实。
Dent病蛋白尿如前所述包含肾小管性蛋白尿和肾小球性蛋白尿成分。(1)对于肾小球性蛋白尿成分治疗:主要借鉴血管紧张素转化酶抑制剂(ACEI)可减轻肾小球疾病和胱氨酸尿症蛋白尿的临床启发,中外学者均有尝试将ACEI用于Dent病蛋白尿的对症治疗中,结果显示临床检测2 h尿蛋白定量虽有轻度减轻,但差异并无统计学意义,也不能改变LMWP的性质[39,40]。在那些误诊为激素耐药性NS的病例中见到,使用环孢素(CsA)或他克莫司(Tac)的病例也可有一定程度的24 h总蛋白定量的减轻,可能与神经钙调蛋白抑制剂(CNI)对Dent病足细胞直接作用有关。或许是因为顾虑CNI慢性肾毒性作用可能加速Dent病肾小管-间质病变的不利影响,未见此类药物的临床研究报道。笔者认为虽然ACEI类不能有效降低Dent病的蛋白尿水平,但该类药物在延缓Dent病肾小管-间质病变慢性进展中可能有益处,尚需长期随访观察。(2)对于肾小管性蛋白尿的治疗:因其是基因突变所致Dent病最基本病生理改变,尚无治疗办法。北京大学第一医院儿科在临床尝试保护营养小管细胞的处理(如大剂量辅酶Q10、1,6二磷酸果糖、百令胶囊等),仅见肾早期损伤指标和24 h尿蛋白定量轻微变化,均无确定疗效。总之,目前对Dent病的蛋白尿尚无有效治疗手段。
治疗特发性高钙尿症的氢氯噻嗪同样可以用于Dent病患者,能有效控制其高尿钙。张宏博和黄建萍[40]采用氢氯噻嗪联合枸橼酸钾治疗15例Dent病 Ⅰ 型男童,观察结果证实噻嗪类药物联合枸橼酸钾治疗可有效控制尿钙排泄,且可防止低钾血症;他们还观察到5例原有的肾钙化和1例肾结石,在治疗后超声复查均完全消失;其中4例在尿钙恢复正常后可停用氢氯噻嗪。北京大学第一医院8例同样使用氢氯噻嗪联合枸橼酸钾溶液治疗高钙尿也取得大致相同的结果,且治疗后无一例有肾钙化或肾结石出现[41]。由此提示:枸橼酸钾溶液配伍氢氯噻嗪治疗Dent病高钙尿症优于氯化钾、补达秀制剂的单纯补钾,在防治因氢氯噻嗪排钾导致的低钾血症同时,还具有因枸橼酸带来的尿液碱化、提升尿枸橼酸盐浓度,防治肾钙化、肾结石的益处[42]。笔者观察到氢氯噻嗪的剂量常常低于其利尿剂量的1/2~2/3即可达到恢复和维持正常尿钙水平的效果,因此,建议初始服药可从小剂量[0.5 mg/(kg·d),1次/d]开始尝试。
总体而言,对于Dent病本身目前尚无有效的特异性治疗手段;对其临床主要症状而言,高钙尿症临床疗效满意,蛋白尿处理尚无确切疗效的治疗方法。
Dent病患者随年龄增长,其肾脏病理改变呈慢性进展趋势,肾功能水平可逐渐降低,部分在成人阶段最终发展至肾衰竭/ESRD。文献报道30%~80%的Dent病患者在30~50岁发展至肾衰竭/ESRD,平均年龄21岁,肾衰竭最小年龄报道为0.5岁[8,43,44]。目前国内已报道的54例Dent病的患儿中(含2例Dent病 Ⅱ 型)无一例因Dent病本身发展导致的肾功能异常;病程中发生的一过性肾功能减退均是由于病程中接受的CNI、ACEI治疗及伴发急性小管-间质肾炎所致。影响Dent病预后的主要因素包括病程、肾钙化程度、肾病理改变(局灶FSGS、局灶球性硬化症、间质纤维化)等。
综上所述,Dent病作为X连锁隐性遗传性肾小管疾病,虽属罕见病,但临床中也时有见到,尤其是对于有LMWP和/或高钙尿伴肾石症的男童,应警惕Dent病,必要时送检CLCN5和OCRL1基因检测以避免误诊和漏诊。儿童期Dent病相对良好,但成人期仍具有肾衰竭风险。因此,儿科医师应积极有效地控制高钙尿症,防治肾钙化和肾石症,尽量避免医源性肾损伤。

























