
分析1例成人型神经元核内包涵体病(NIID)患者的临床、影像学及皮肤病理学特征,探讨成人型NIID的临床、病理特点及诊断方法。患者为61岁女性,临床表现为进行性认知功能减退、发作性意识障碍、卒中样发作、发作性消化道症状;头颅磁共振弥散加权像显示皮髓交界绸带样分布高信号,持续存在。皮肤活体组织检查(活检)病理发现脂肪细胞、纤维细胞及汗腺细胞核内嗜酸性核内包涵体。该病例提示成人型NIID是具有高度临床异质性的慢性神经变性病,头颅MRI弥散像皮质下绸带征及皮肤活检示核内嗜酸性包涵体有助于诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
神经元核内包涵体病(neuronal intranuclear inclusion disease,NIID)是一种慢性进展性神经系统变性疾病,病理特征为神经元、内脏器官细胞核内存在嗜酸性透明包涵体[1]。NIID的临床表现具有高度异质性,主要包括锥体系和锥体外系症状、小脑性共济失调、痴呆、痫性发作、周围神经病和自主神经功能障碍等,散发和遗传性病例均见于报道[1]。自从研究证实皮肤活体组织检查(活检)在NIID诊断中的价值后,国外文献关于NIID的报道尤其是成人型NIID的报道数量逐渐增多[2]。经文献检索,国内目前尚未见相关报道。我们报道1例以发作性意识障碍、卒中样发作、智能减退及发作性消化道症状为主要临床特征的NIID患者,分析其临床特点、影像学变化、皮肤病理改变,并复习相关文献,旨在帮助临床医生提高对NIID的认识。
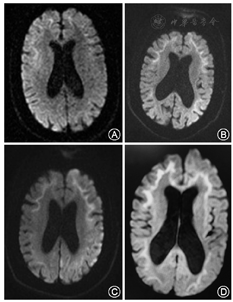
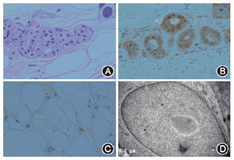
患者女性,61岁,家庭主妇。主因"反复发作性意识模糊7年,再次发作4 d"于2017年3月20日收入我院。患者于2010年2月1日无明显诱因出现意识模糊、胡言乱语,不能与人沟通,不伴发热、肢体无力、肢体抽搐等,症状持续约1 h后消失,当时检测血糖水平在正常范围。2 d后出现剑突下胀痛,反复非喷射性呕吐,呕吐物为胃内容物,出现流涎,我院消化内科给予胃黏膜保护剂、止吐药、胃肠动力药物等对症治疗,仍然间歇性上腹疼痛和呕吐,症状持续2个月逐渐好转。2010年6月开始患者出现反复发作性意识模糊,不能识别家人和朋友,无法言语沟通,每年发作2~3次,症状类似于2010年首次发作,每次持续半小时至数小时不等,经补液或自行好转。2016年2月及8月分别出现间歇性上腹痛、呕吐及流涎,症状波动迁延达2个月余,逐渐自行缓解。2017年3月16日患者再次出现意识模糊,并出现言语不能,右侧上、下肢活动较左侧显著减少,尿潴留。无发热、肢体抽搐和活动障碍等。家人发现自2010年患者记忆力逐渐减退,以近记忆力下降显著,如反复询问刚刚发生的事情,不能完整做完一项任务等,发作性症状间期能够从事简单的家务活动。患者于2012年行"胆囊切除术"史。无高血压、糖尿病、心脏病、肝炎及结核史,家族中无类似病史。入院检查:心、肺未见异常,腹壁软,肝脾未触及肿大,肠鸣音正常。嗜睡,意识内容减少,混合性失语,神经系统检查不能配合,双侧瞳孔等大等圆,对光反应存在,眼球运动充分,右侧肢体主动活动较左侧减少,疼痛刺激可见右上下肢肌力小于左侧。共济运动、深浅感觉检查不能配合。四肢腱反射减低,双侧掌颏反射和吸吮反射阳性,双侧巴宾斯基征阴性。颈软,脑膜刺激征阴性。辅助检查:甲状腺功能及甲状腺过氧化物酶抗体、肿瘤标志物、免疫全套、抗中性粒细胞胞质抗体、血氨、血乳酸、肌酸激酶及同工酶等均在正常范围。脑脊液常规及生化检查结果正常,脑脊液和血自身免疫性脑炎及副肿瘤相关抗体均为阴性。尿有机酸及血脂酰肉碱及氨基酸检测未见异常。腹部X片及腹部CT未发现肠梗阻,胰腺MRI平扫及增强未发现胰腺占位,胃镜显示慢性非萎缩性胃炎。神经传导检测与针极肌电图检查结果示:上下肢运动和感觉神经传导速度检测均在正常范围,F波出现率、潜伏期均正常;皮肤交感反应(SSR)检测:双侧上、下肢SSR均未引出波形。左肱二头肌活检病理检查:肌纤维大小形态正常,肌质和核内未见异常物质沉积,改良Gomori三色染色未见不整红边纤维。线粒体DNA热点突变位点(mtDNA-3243、3271、3260、8344、8356和8993)筛查均为阴性。头颅MRI动态变化:2010年2月4日弥散加权成像(DWI)可见双侧额叶沿皮髓质交界处高信号影;2017年3月19日DWI可见双侧额、顶、颞、枕叶沿皮髓质交界处对称性绸带状高信号影,左侧额、顶、颞、枕叶皮质多发斑片状高信号影;2017年3月23日DWI可见双侧额、顶、颞、枕叶沿皮髓交界处分布对称性绸带状高信号影,左侧额、顶、颞、枕叶皮质多发斑片状高信号影,皮质区病灶较2017年3月19日范围略扩大;2017年4月20日DWI可见双侧额、顶、颞、枕叶沿皮髓交界处分布对称性绸带状高信号影,与前2次比较,左侧额、顶、颞、枕叶皮质病变大部分消失(图1)。皮肤病理改变:HE染色可见部分汗腺腺体细胞、脂肪细胞和纤维母细胞细胞核内存在嗜酸性核内包涵体,免疫组织化学染色可见包涵体泛素和p62阳性。电镜可见汗腺腺体细胞、脂肪细胞和纤维母细胞的细胞核内类圆形包涵体,大小约1.5 μm×5 μm,由纤维样物质构成,周边有晕(图2)。治疗及病情演变:入院后给予对症支持治疗,第4天患者意识转清,检查发现存在混合性失语。入院后第2天右侧肢体肌无力进行加重,并于第7天进展至右侧肢体肌力0级,第11天开始右侧肢体肌力逐渐恢复,并开始能听懂他人言语。第19天出现上腹部疼痛,反复呕吐,流涎,消化道症状与2010年及2016年2次发作完全一致,持续20 d左右症状消失。入院后第45天,右肢肌力恢复至Ⅳ级,运动性失语。
该患者临床主要表现为发作性脑病,发作性消化道症状,缓慢进展的智能减退,自主神经损害;头颅MRI DWI显示持续存在的皮髓质交界处高信号,皮肤活检发现汗腺细胞、脂肪细胞和纤维细胞胞核内嗜酸性包涵体,p62和泛素免疫组织化学染色阳性。一系列临床和实验室检查可以排除线粒体脑肌病、狼疮性脑病、桥本脑病及低血糖脑病等,散发型成人型NIID诊断成立。
NIID是一种罕见的神经变性病,1968年Lindenberg等[3]首先报道1例28岁男性患者,儿童期起病,主要表现为行为异常和缓慢进展的多种神经系统症状,包括不自主运动、共济失调及自主神经损害等,脑组织病理活检显示神经元核内嗜酸性包涵体。20世纪80年代,Sung等[4]报道1例21岁临床与病理相似的女性患者,病理发现中枢神经系统广泛分布的核内包涵体,将并之命名为NIID,并推测其病因可能与特殊的病毒感染及遗传易患性有关[2]。2003年Takahashi-Fujigasaki[2]根据发病年龄将NIID分为婴儿型、青少年型及成人型,根据遗传方式分为散发型与家族型。
NIID的病理生理机制尚不明确。首先,NIID的核内包涵体病理意义未明。在NIID患者中,核内包涵体经常存在于形态上基本正常的神经元。即使病理检查存在广泛的核内包涵体,也并不一定伴随神经元变性、丢失。核内包涵体并不一定有细胞毒性,NIID的核内包涵体含有泛素-蛋白酶体蛋白降解系统的一些蛋白,如泛素和p62,其形成与泛素蛋白酶体蛋白降解系统的功能障碍有关。有研究提示NIID的核内包涵体形成可能与多聚谷氨酰胺疾病具有相似的病理生理途径,是由三核苷酸重复序列(胞嘧啶-腺嘌呤-鸟嘌呤,编码谷氨酰胺)特异性基因扩增引起的神经退行性疾病[1]。多聚谷氨酰胺疾病的核内包涵体是由含有异常谷氨酰胺片段的疾病相关蛋白聚集形成。研究发现NIID中的神经元核内包涵体对1C2(一种识别异常扩增的聚谷氨酰胺的单克隆抗体)具有免疫反应性,并且含有ataxin 3等含有多聚谷氨酰胺的蛋白。但是目前还没有发现NIID由多聚谷氨酰胺异常扩增所致的直接证据[5,6]。
散发型NIID多起病于51~76岁,病程1~19年不等。症状多样,根据受累部位可分为中枢神经系统、周围神经、自主神经受累3组症状[7]。中枢神经系统受累症状包括:痴呆(94.7%),为其首发和核心症状;共济失调(52.8%);发作性意识障碍(39.5%),意识障碍轻重不一,持续时间数小时至数天不等;行为异常(26.3%),表现为易怒、言语不清、失用症、沉迷于赌博等;亚急性脑炎样表现(21%),表现为发热、头痛、呕吐、意识障碍,晚期可出现无动缄默征;其他如强直(23.7%)、震颤(18.4%)、全面强直发作(13.2%)。类似本例卒中样发作亦见报道[8]。周围神经受累表现:感觉障碍(28.6%),轻微震动觉减退,末梢型肢体麻木;肌力下降(27%),四肢远端肌力轻度下降;自主神经受累表现:瞳孔缩小(94.4%)、膀胱功能障碍(尿失禁;33.8%)、呕吐(15.8%)、晕厥(8.1%)。家族型NIID临床表现与散发型相似,但症状发生的比例略有不同。根据其症状可分2个亚组:痴呆组及肢体无力组:痴呆组发病年龄在40岁以上,以痴呆为首发及核心症状,可伴轻微的自主神经功能紊乱、亚临床周围神经病变;肢体无力组:发病年龄16~39岁,以下肢无力为首发症状,逐渐出现感觉障碍和自主神经功能障碍。病程到20年左右可出现痴呆与白质病变。
NIID患者头颅MRI T2WI和FLAIR序列可出现明显的脑白质病变,这些白质病变在T2WI双侧对称并相互融合,T2WI高信号的区域在T1WI呈低信号。DWI可出现特征性沿皮质、髓质交界处高信号,病灶随着疾病进展不断向后延伸[9]。正如本例患者2010年的MRI表现,DWI高信号病灶仅存在于额叶皮髓质交界处。随着病程进展,至2017年,DWI高信号病灶从额叶向顶枕叶延伸。即使病程到了无动性缄默晚期,T2WI出现广泛的脑白质病变时,DWI高信号也不会累及深部白质。DWI沿皮质、髓质交界处高信号改变,是NIID特征性影像表现,在散发型病例中的出现率为100%,而在家族型病例中的出现率为(81.8%)[7]。DWI的这种特征性影像学改变尚未见于其他疾病的报道,该影像学特征对于诊断NIID具有很高的提示价值,我们将这种特征性影像命名为皮质下绸带征(subcortical lace sign)。
有研究[10]显示在NIID患者的中枢神经系统神经元和星形胶质细胞中存在嗜酸性核内包涵体,也可见于外周神经系统以及除骨骼肌、肝细胞外的很多器官体细胞。神经元包涵体位于核内,直径为1.5~10.0 μm,泛素和p62阳性,电镜下核内类圆形无膜结构,由纤维样物质构成。近年来相关研究提示NIID患者皮肤的脂肪细胞、纤维母细胞、汗腺细胞也存在嗜酸性核内包涵体[9],直径1.5~3.0 μm,皮肤有关细胞的核内包涵体的光镜和电镜下特征与中枢神经系统内相同。有文献报道脆性X相关震颤共济失调综合征(FXTAS)患者可表现为痴呆和脑白质病变,并且在这些患者的神经元、胶质细胞和体细胞内可观察到和NIID类似的嗜酸性泛素化阳性的核内包涵体[11]。目前尚无FXTAS核内包涵体皮肤病理或电镜的相关文献报道,且FXTAS也没有NIID的特征性影像学改变。因此,皮肤活检存在特征性核内包涵体可作为NIID的主要诊断标准。
NIID属于罕见病,目前尚没有大宗的临床试验研究,文献报道较少。部分研究报道在表现为亚急性脑炎的病例中,激素冲击治疗对于短期内减轻脑水肿、改善意识状态可能有效[7]。由于激素治疗NIID的病例数量有限,激素长期治疗的有效性仍有待商榷。
无
None declared

























