
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
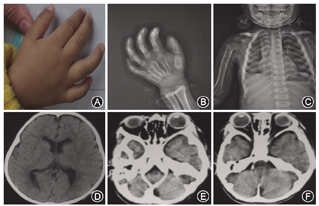
例1 患儿女性,3岁。因双眼流泪半年、加重伴视力显著下降2周,于2012年12月收入解放军兰州总医院。患者自幼双眼视力低于同龄儿童,曾经于2011年3月就诊当地医院,诊断为双眼角膜发育不良;药物治疗无效。1年前因生长发育迟缓及双手畸形于本院儿科诊断黏多糖贮积症1型(Hurler-Sheie综合征)。入院检查:患者一般情况良好,体重及发育情况与同龄儿无显著差异。四肢长骨远端膨大;胸部X线提示右侧肱骨上端略宽大,外科颈骨皮质不光滑,余骨质密度正常,未见骨质破坏及骨折征象;右侧尺挠骨远端干骨骺端宽大,略毛糙,见第1~5掌骨骨皮质扭曲;颅脑CT提示幕上脑室扩大(图1)。眼部检查:视力右眼0.05,左眼0.01,双眼不接受矫正;眼压右眼33mmHg(1 mmHg=0.133 kPa),左眼29 mmHg。双眼角膜全层中度混浊,角膜内皮轻度水肿,A超角膜测厚490~510 μm;前房轴深1.5~2.0 CT,虹膜纹理欠清晰,自然光线下瞳孔3~4 mm,对光反应略迟钝。患儿家长拒绝UBM检查。入院诊断:双眼继发性青光眼;黏多糖储积症。治疗策略:患者晶状体前后径显著增大,因此,给予患者硫酸阿托品眼用凝胶涂眼治疗,改善睫状肌张力。并给予布林佐胺滴眼液及盐酸贝他洛尔滴眼液点眼治疗,患者经过治疗后,眼压右眼20~24 mmHg,左眼19~23 mmHg,双眼角膜水肿改善;前房深度及眼轴长度始终无明显变化。
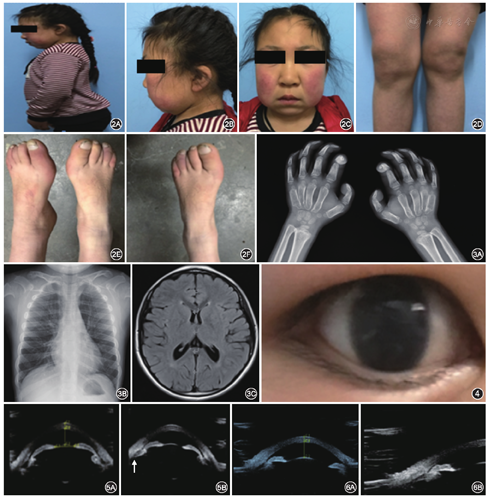
例2 患儿女性,11岁。因双眼视力差5年余,于2015年6月收入解放军兰州总医院。患儿自幼双眼视力低于同龄儿童,曾经于2005年就诊当地医院,诊断为双眼角膜发育不良;药物治疗无效。既往史:1年前因发育滞后我院儿科诊断为黏多糖贮积症1型。入院全身体检:身高110 cm,体重25 kg,颜面及颈项部均表现为典型的颅骨远端肥大,关节活动能力下降,颌面关节功能下降;脊柱前屈;而双侧手足关节及膝关节均表现关节远端肥大,功能下降,行走呈跛行步态(图2)。胸部X线提示右侧肱骨上端略宽大,外科颈骨皮质不光滑,余骨质密度正常,未见骨质破坏及骨折征象;右侧尺挠骨远端干骨骺端宽大,略毛糙,见第1~5掌骨骨皮质扭曲;颅脑CT提示幕上脑室扩大(图3)。患儿就诊时就读小学2年级,成绩中等,心理门诊及神经内科会诊提示:患者智商测量值105~120,智商低于同龄儿童,能胜任一般学习任务;儿科及内分泌科会诊意见:黏多糖贮积症1型。入院眼部检查:视力右眼0.12,左眼0.1,双眼不接受矫正;眼压右眼45 mmHg,左眼38 mmHg。双眼角膜全层中度混浊,角膜内皮皱褶(图4),前房轴深2~3 CT,虹膜纹理欠清晰,自然光线下瞳孔直径约3 mm,对光反应迟钝。应用UBM(图5)及眼球A超、角膜厚度等生理指数测量,患儿角膜厚度为0.537~0.562 mm,且角膜混浊水肿对Goldman压平式眼压测量结果影响并不大。双目眼底镜眼底检查模糊可见视盘及大血管,C/D为0.4~0.5,但很难拍照。由于儿童屈光间质混浊明显,其视觉电生理检查虽然表现为波幅下降及潜时延长,但不能单独证明视神经损伤情况,因此未予列出。入院诊断:(1)双眼继发性青光眼;(2)黏多糖贮积症。患者睫状体前位显著,周边房角狭窄但无明显关闭,小梁网结构范围密度增大,因此,给予患者硫酸阿托品眼用凝胶涂眼治疗,改善睫状肌张力。并给予布林佐胺滴眼液及盐酸倍他洛尔滴眼液点眼治疗,患者经过治疗后,眼压右眼(17.13±3.11)mmHg,左眼(16.09±2.67)mmHg,双眼视力提高,右眼0.25 2.50 DS=0.5,左眼0.2
2.50 DS=0.5,左眼0.2 1.75 DS=0.4;膜水肿改善。治疗过程中,瞳孔始终无明显散大,前房深度及眼轴长度无明显改变。给予患者硫酸阿托品眼用凝胶涂眼后,眼前节UBM提示,患者双眼晶状体厚度降低,晶状体虹膜隔范围缩小,而前房角无明显进一步狭窄表现(图6),眼压降至15~19 mmHg。
1.75 DS=0.4;膜水肿改善。治疗过程中,瞳孔始终无明显散大,前房深度及眼轴长度无明显改变。给予患者硫酸阿托品眼用凝胶涂眼后,眼前节UBM提示,患者双眼晶状体厚度降低,晶状体虹膜隔范围缩小,而前房角无明显进一步狭窄表现(图6),眼压降至15~19 mmHg。
黏多糖贮积症是一种先天性风湿病。Ⅰ型患者体征及症状表现为最典型,是黏多糖贮积症的原型,常染色体隐性遗传。由于溶酶体a-L-艾杜糖醛酸酶缺乏,使黏多糖分解发生障碍,体内各种组织细胞内有分解不全的黏多糖沉积,并随尿排出体外。在内脏病变、骨骼畸形和智力障碍方面的症状都很严重[1,2]。骨关节严重畸形,手指粗短,短颈,脊柱后弯。各关节逐渐挛缩强直,手指固定于半屈位,呈爪状,身材矮小。
早期在黏多糖储积症患者并发症研究中,通常认为青光眼是该病的早期并发症,主要源于储集的黏多糖导致小梁网填塞和组织变性扭曲,同时发生角膜及虹膜、晶状体囊膜的细胞外基质黏稠变性[3],表现为病理发病过程中眼压持续升高,可检测的指标证实了视功能损伤。黏多糖储集于眼部的主要症状也可能源于虹膜和晶状体囊膜细胞外基质增生及性状改变,从而发生晶状体虹膜隔阻滞[4],因此,通过药物治疗解除相对解剖阻隔[5],可望改善高眼压状况。回顾以往病例研究,通常认为房角组织导致的急性闭角型青光眼发病的可能性非常低[6]。但是,临床治疗的中心要点依旧集中于防止眼内压失控[7]。较长时间的观察证实,5岁以后患儿的青光眼发病率提高,间接证实黏多糖储积导致的细胞外基质性状改变与眼压之间的相关性[8],而机械性阻滞作用可能在早期得到了部分代偿。成年患者的角膜病理检查证实,在上皮细胞和内皮细胞和角膜基质中,存在酸性黏多糖颗粒[9],从而为细胞外基质变性理论提供了客观依据。
本组例1患儿发病早,病情较重,经过治疗后眼压初步控制,患者视力基本保持,但由于全身一般情况较差,随访6个月后失访;例2患儿经过密切随访治疗,迄今视力矫正视力保持0.6~0.8,但由于患者不能配合视野检查,仅能粗测周边动态视野范围大致正常,不能详细检查静态视野视阈值,期待进一步治疗后,伴随患儿智力进一步发育,能够配合视野检查。
黏多糖储积症的早期往往表现为角膜水肿混浊,视力下降。幼儿由于眼压症状及体征不显著而忽略,但是,最终青光眼症状逐渐明显,表现为视力及视野损伤[10]。在早期研究中,由于患者早期死亡比例较大,青光眼损伤往往被忽略[11]。
在青光眼发病之初,由于角膜混浊及异常增高的组织细胞张力,患儿的眼压显著增高,尽管往往表现与实际眼内压不相一致,但是,针对角膜厚度的检查和降眼压治疗后患儿视功能改善[12],能够证明角膜混浊同时发生的晶状体-虹膜隔阻力增大导致的继发性青光眼性质的结构改变,已经影响了患儿的视功能。因此,早期规范的抗青光眼降眼压治疗是必须的。
长期大数据临床观察证实,在发病过程中,患儿的眼压表现为不可逆的逐步升高过程,其疑似及确诊青光眼比例远远高于实际确诊数量。继发性青光眼的诊断,主要依赖于角膜厚度、前房角、视功能的随访频度,随着进行性损伤进展,继发性青光眼的诊断数量不断增加,最终临床确诊青光眼的数量远远高于角膜症状出现时的确诊患者数量。因此,角膜厚度与高眼压互参、诊断性降眼压治疗及患儿视功能修复过程,均可作为青光眼的诊断策略。并且,结合全身情况与视功能损伤特征,可以与生理性大角膜、发育性青光眼相鉴别。
本组2例患儿均为内分泌科、儿科及神经内科确诊病例,病情稳定期由于视力下降就诊。后者年龄较大,可以配合UBM检查,从而得到指导临床治疗的客观检查结果。但患儿临床观察时间尚短,目前对于治疗效果不能做出进一步论断。

























