
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
基于二代测序技术定义的克隆性造血(Clonal hematopoiesis,CH)概念为血液病临床诊治带来许多新的认识,也引起很多不解和困惑。CH的临床诊断问题需要科学的方法论和视角,我们从造血干细胞(Hematopoietic stem cell,HSC)生物学角度来科学阐述CH。
CH是指具有分子遗传学突变特征的HSC,通过多系造血分化,形成携带重现性生物学标志的终末分化成熟血细胞。因此,CH是一种非恶性扩增的造血模式,具有竞争性克隆优势和多系造血分化成熟能力两大核心特征。
CH的发生是HSC内在衰老和外在环境因素阳性筛选共同作用的必然结果和客观存在。DAN复制中心法制决定了HSC自我更新过程中会不断积累DNA复制错误,每一个HSC每10年随机积累(1.3±0.2)个DNA碱基错配。如图1所示,HSC衰老和多种环境因素共同形成了CH。
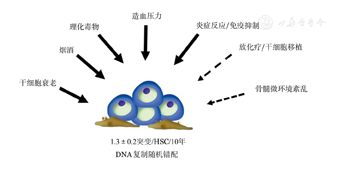
CH是具有独特生物学特征的HSC造血模式:①CH具有竞争性克隆扩增优势。分子遗传学突变是克隆扩增的内在驱动力,外界环境因素的压力性选择促进克隆扩增。②CH保持多系造血分化能力。这是CH区别于恶性克隆扩增、分化成熟阻滞最本质的不同。③CH多系造血分化能力不平衡。呈现髓系分化偏颇,易积累多次突变并进展为髓系恶性克隆。④CH髓系偏颇分化形成的终末成熟细胞具有病理性。TET2突变的单核-巨噬细胞通过NLRP3炎症小体活化、多种趋化因子以及多种炎症因子加速冠状动脉粥样硬化形成。⑤CH有较高风险克隆演变为髓系恶性克隆,CH者髓系肿瘤发生风险高达11~12倍。但是,伴有CH的健康人群最终发生髓系肿瘤的比例极低(约4%),年发生风险概率0.5%~1%。CH克隆发展的主流模式是非病理性、非白血病性的稳定共存模式,仅极少数CH可进展为白血病克隆(图2)。
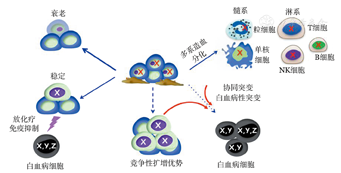
携带白血病相关基因突变并具有多系造血分化成熟能力的CH定义为白血病前期干细胞(pre-leukemia stem cell,pre-LSC)。pre-LSC属于功能性HSC,并非病理性白血病细胞/白血病干细胞。但pre-LSC具有恶性克隆演变潜能,是白血病发生的克隆起源。CH的分子遗传学突变作为驱动/起始突变,克隆演变为白血病克隆需要以下三种必要且充分条件共同作用:①克隆起源:驱动突变的基因类型及分子特征,使得pre-LSC具有高度的竞争性克隆扩增优势。如DNMT3A R882、TP53、IDH2、RUNX1和RNA剪接子突变等。②克隆筛选:外界环境因素发挥阳性筛选作用促进克隆扩增,如放化疗、免疫抑制、炎症反应等。③克隆突变积累:克隆扩增过程中获得有效的多次协同突变(白血病性突变),如NPM1c、FLT3-ITD/TKD、NRAS/KRAS等。通过以上病理机制,pre-LSC获得恶性增殖能力,丧失了终末分化成熟能力,克隆演变为白血病干细胞(图2)。
CH对于AML诊断、治疗及预后评估中有以下几个问题需要厘清:
1.AML诊断时分子遗传学突变筛查需要甄别CH分子突变与白血病性突变,前者不能作为微量残留病监测的生物学标志。常见的CH分子突变种类有表观遗传学(DNMT3A、TET2、IDH1/2、ASXL1等)、RNA剪接子以及Cohesins蛋白等,白血病性突变主要是信号通路激酶和转录因子突变,如NPM1、FLT3-ITD/TKD、NRAS/KRAS、KIT等。研究报道30%~70%的初诊AML有CH分子突变,治疗完全缓解(CR)后这些突变多数持续存在,并且化疗具有促进非白血病性CH克隆扩增作用。因此,缓解后这些非白血病性CH分子突变负荷增加不是预示AML复发的分子标志。
2.CH对AML缓解率、无病生存/总生存、复发及耐药的临床意义各有侧重。CH分子突变定性结果不影响AML诱导缓解率,不影响CR后异基因造血干细胞移植的无病生存/总生存;但是诊断时CH分子突变高负荷定量分析是无复发生存(HR=2.99)/总生存(HR=3.30)的危险因素。AML CR后近期复发主要克隆来源是白血病性克隆,而pre-LSC是白血病缓解5~10年后复发时白血病重建的重要克隆起源;2017 ASH报道DTA(DNMT3A、TET2、AXSL1)突变阳性AML患者缓解后非DTA突变持续阳性是独立预后因素,5年累积复发风险高。DNA甲基化(如DNMT3A)、组蛋白修饰(如EZH2)突变会导致白血病细胞耐药,是AML预后不良因素。DNMT3A R882突变的AML通过染色质重构能力损伤而对标准剂量蒽环类药物耐药,提高蒽环类药物剂量可改善耐药;EZH2突变的白血病细胞系通过H3K27me3水平下调导致HOX等干性基因和耐药基因表达,产生阿糖胞苷耐药,体外经蛋白酶体抑制剂作用上调EZH2表达可减轻耐药程度。
3.CH是治疗相关AML/MDS(t-AML/MDS)发生的克隆起源。既往认为t-AML/MDS是实体瘤患者放化疗治疗措施诱导HSC产生新的恶性突变所致,目前研究证实20%~30%的实体瘤患者发病时即已存在CH。病例-对照研究表明,伴有CH的实体瘤患者高风险发生t-AML/MDS(OR=5.75),中位发生时间5.6年;队列研究发现,CH是实体瘤患者发生t-AML/MDS的高危因素(HR=14.0),5年累积发生率为30%。t-AML/MDS主要病理机制是放化疗作为阳性筛选压力促使本已存在的CH克隆扩增、积累突变后克隆演变为髓系肿瘤。病理机制认识的更新对于淋巴瘤等实体瘤患者制定合理的干细胞移植策略有指导意义。大样本队列研究证实伴有CH的淋巴瘤患者自体造血干细胞移植(auto-HSCT)后10年t-AML/MDS累积发生率明显增高(14.1%),并且总体生存率(30.4%)较差。高龄淋巴瘤患者行HSCT时宜做自身CH筛查,并权衡异基因和自体供者的风险。
异基因HSCT治疗恶性血液病,发生植入不良或者受者不明原因血细胞减少时,需要考虑到供者植入型CH克隆演变风险,特别是对于伴有高风险白血病进展潜能的CH分子突变(如DNMT3A、TP53、IDH2、RUNX1和RNA剪接子突变等)或者有2种及以上髓系肿瘤相关基因突变的50岁以上供者,应考虑供者型白血病发生的风险。因此,自体或异基因HSCT供者筛选应考虑供者是否伴有CH及其分子突变特征,有效降低移植后造血重建不良的风险。
再生障碍性贫血(AA)与MDS鉴别诊断是临床难点问题。MDS的生物学本质是起源于HSC的克隆性髓系肿瘤,AA是T细胞免疫介导的HSC衰减,二者干细胞生物学本质不同。约80%的MDS患者存在至少一种髓系肿瘤基因突变,而近2年研究发现近50%的AA患者也存在CH,约1/3的患者具有MDS相关的基因突变。
科学解读获得性骨髓衰竭症CH分子突变的鉴别诊断意义需要把握以下两个关键点:①AA和MDS都存在CH,但导致临床病理的因果关系不同。MDS是一组干细胞恶性克隆性演变异质性疾病,CH做为驱动突变是MDS临床表型的恶性克隆起源;T细胞始动免疫打击HSC导致了AA临床表型,CH是HSC在免疫打击后阳性/压力性筛选的结果,AA患者伴有的CH有克隆扩增优势,有克隆演变为MDS/AML的风险。② CH对于AA和MDS临床诊断意义的侧重点不同。MDS诊断的基石是骨髓形态发育异常,特征性染色体异常也可以作为有力的证据。CH分子突变可做为MDS诊断的补充证据,而不宜做为唯一诊断依据。而AA诊断和鉴别诊断的核心是排除其他导致骨髓衰竭的血液和非血液系统疾病,积极寻找自身T细胞免疫打击的证据,如HLA-Ⅰ类抗原位点缺失、6号染色体单亲二倍体、PNH克隆等,存在CH目前尚未纳入AA诊断和鉴别诊断体系中。
AA、MDS的CH诊断问题解惑之道在于:对于AA,我们应着眼于CH对AA免疫抑制治疗的预后指导意义,动态监测克隆演变,及早评估是否进行HSCT治疗干预;对于MDS,我们应着眼于CH对于MDS克隆的发生、发展及白血病演变风险的预后意义,明确MDS诊断后,再根据突变特征评估预后风险,制定科学的治疗策略。
CH是HSC内在衰老机制和外在环境选择性压力阳性筛选共同作用的客观存在,是多种血液系统疾病发生的病理基础。科学认识CH生物学特征,辨析不同疾病中CH的临床诊断应用价值和侧重点,用科学的方法论指导我们认识复杂的临床问题,诸多困惑迎刃而解。

























