
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
血栓性微血管病(TMA)是由各种原因所致的一组以微血管病性溶血性贫血(MAHA)、血小板减少、缺血性器官受累为特征的急性临床病理综合征。其病理及临床表现相似,而发病机制多样,疾病谱涉及肾脏病、风湿、血液、产科及肿瘤等专业领域,诊断和治疗较复杂。近年,随着对TMA发病机制研究的不断深入,其诊疗方案有了较大突破,但仍存疑惑。本文拟从临床实用角度对TMA研究进展作一梳理归纳,其中重点叙述非典型溶血尿毒综合征(aHUS)和血栓性血小板减少性紫癜(TTP)。
以往TMA临床分为两类:溶血尿毒综合征(HUS)和TTP。近年来,随着对TMA发病机制的深入了解,TMA分类及其诊疗思路发生了很大的变化。Kato等[1]将TMA分为TTP、典型HUS、补体相关aHUS、继发性TMA。最近Brocklebank等[2]则将TMA分为原发性TMA、感染相关TMA和继发性TMA,其中原发性TMA又分为遗传性和获得性TMA,前者因基因突变致病,包括原发性aHUS、遗传性TTP等,后者存在自身抗体,包括继发性aHUS、继发性TTP。感染性TMA主要由各种感染所致,包括出血性大肠杆菌、肺炎球菌分泌毒素所致的HUS等[2]。另有学者提出药物,自身免疫性疾病,妊娠,感染,肿瘤,溶血、肝酶升高、血小板减少综合征(HELLP),器官移植与肾小球肾炎等疾病均可在特定条件下触发TMA,按发病机制其中一部分可归类到HUS或TTP,但仍有一部分机制尚不清楚,故而主张将其归类至"其他因素所致TMA"[3]。梳理相关文献,将TMA的分类按病因整理归纳,见图1。
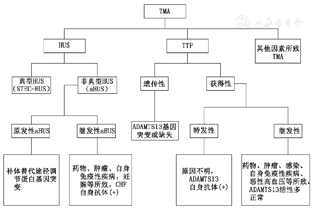
注:TMA:血栓性微血管病;HUS:溶血尿毒综合征;TTP:血栓性血小板减少性紫癜;CHF:补体H因子;ADAMTS13:金属蛋白酶域蛋白家族13号成员
HUS是以MAHA、血小板减少、急性肾衰竭"三联征"为特征的一种TMA[4]。以往根据有无腹泻将HUS分为腹泻HUS和无腹泻HUS,后有研究发现25%~30%的aHUS患者有腹泻,故该分类方法已被弃用。近年,学者们发现补体替代途径相关调节基因突变或存在补体H因子(CFH)自身抗体是HUS的新致病因素,主张更新HUS的命名、分类与诊疗方案,目前临床上通常将HUS分为典型HUS与aHUS两种[1,5]。
典型HUS儿童多发,大多与腹泻相关,预后较好[1]。主要发生在产志贺毒素(Stx)的出血性大肠杆菌(STEC)导致的肠道感染后5~7 d(血清型主要为O157:H7或O104:H4),故典型HUS也称为STEC-HUS[6]。其诊断主要依据为:最近2周有腹泻病史,"HUS三联征",大便细菌培养STEC阳性或血清Stx检测阳性[7]。典型HUS为一自限性疾病,治疗主要为对症支持处理,血浆置换(PE)和抗补体治疗的疗效不确切,急性期抗生素治疗可能加重病情[1,8]。
aHUS与典型HUS同属HUS,但二者在发病机制、临床特征及预后等方面均有较大差别。aHUS是近年TMA研究较热的一种疾病,研究进展较多。
关于aHUS的定义及分类一直存在争议,随着对该疾病发病机制的深入研究,目前大部分学者认为除典型HUS以外所有的HUS都可纳入aHUS(广义),也有学者主张只能将补体相关HUS称为aHUS(狭义)[1,5,7]。aHUS可分为原发性aHUS、继发性aHUS,一些指南所述aHUS主要是指补体介导的原发性aHUS[1,5,6]。
补体系统由补体固有成分、补体调节蛋白、补体受体成分三部分组成。其活化主要包括经典途径、凝集原途径和替代途径,C3转换酶是这三条补体活化途径形成终末共同通路的交汇点,并顺序激活下游其他补体因子,生成C5转换酶,继而生成攻膜复合物,发挥补体效应。
补体调节蛋白包括血浆蛋白和膜复合蛋白,在替代途径中前者包括补体H因子(CFH)、补体I因子(CFI)等,后者包括CD46等。正常生理情况下,补体调节蛋白抑制替代途径活化或加速C3转换酶的水解。替代途径调节蛋白或其补体固有成分基因突变,可导致补体替代途径的持续过度活化,产生过多的攻膜复合物和C5a,攻击自身正常的细胞或器官,造成相应组织器官功能障碍[7]。
aHUS主要由补体替代途径的调节基因(如CFH、CFI、CD46)及补体固有成分(B因子、C3)突变或CFH自身抗体所致病,这些基因突变或CFH抗体使补体调节蛋白对替代途径活化的抑制作用减弱或补体固有成分功能上调,从而导致补体系统过度活化,诱导血管内皮细胞损伤和血小板聚集[5,7]。最新研究发现补体H因子相关基因(CFHR)突变编码的FHR蛋白能竞争结合CFH,从而影响CFH对替代途径的调节[9]。研究还发现,在aHUS患者中编码血栓调节蛋白(THBD)和编码二酰基甘油激酶ε(DGKE)存在基因突变,二者是凝血途径调节基因。THBD除了起重要的抗凝作用外,还能结合C3b或CFH使其失活,故THBD缺失也可致补体替代途径异常活化。DGKE基因突变通过介导内皮细胞胞内信号传递,损伤内皮细胞,其细胞膜上的CD46表达下降,但不直接影响补体系统的活性[10,11]。
基于目前的指南与共识[1,5,8],aHUS诊断的基本思路为在诊断TMA的基础上,排查TMA其他类型及病因,同时检测补体调节因子和裂解产物,寻找aHUS发病的直接证据。确诊依靠补体系统血清学依据和基因检测。(1)排除TMA其他类型(凡疑诊aHUS者均应排查):①典型HUS:粪便培养、检测血清Stx和抗脂多糖抗体;② TTP:测定血清金属蛋白酶域蛋白家族13号成员(ADAMTS13)活性(<10%);ADAMTS13抗体(+);③继发因素:自身免疫性疾病、肿瘤、妊娠、感染、药物、移植后等。(2)补体系统异常的证据:①血清补体C3下降、C4正常,常表明补体替代途径活化;②CFH、CFI、B因子水平及活性下降;外周血单个核细胞CD46水平下降;③CFH、CFI、CD46、CFHR、B因子、C3等基因突变;④CFH自身抗体检测(+);以上检测指标阳性支持aHUS诊断,但结果阴性不能排除aHUS。
(1)血浆治疗:从20世纪80年代开始至今,血浆治疗一直是aHUS的主要治疗方法。血浆治疗方式包括PE或血浆输注(PI)。血浆治疗的目的是清除异常的补体调节蛋白和CFH抗体等致病因子,同时补充正常的补体调节蛋白[2]。多个指南[1,5]推荐凡疑诊aHUS者应立即行PE或PI。连续PE或PI治疗效果较间断PE或PI效果更佳;也可连续治疗3~5 d后,根据TMA症状和实验室指标调整PE治疗频率。若持续PE 5 d后,血小板计数、乳酸脱氢酶、血肌酐无改善趋势,表明PE效果治疗差,应考虑转而使用依库珠单抗(eculizumab)治疗[7]。对继发于免疫因素及CFH抗体(+)的患者,在进行PE或PI的同时通常还需加用糖皮质激素或免疫抑制剂[1]。CD46为PE不能清除的膜蛋白,故CD46突变导致的aHUS行PE治疗无效[7,8]。(2)补体抑制剂治疗:依库珠单抗为重组人抗补体C5单克隆抗体,能竞争性阻断C5裂解为C5a和C5b,抑制补体终末阶段的活化,减轻炎症反应和内皮损伤[8]。临床试验证实依库珠单抗能有效抑制补体介导的aHUS,改善aHUS患者的肾功能和生活质量,且不良反应少[2,5]。日本等国指南推荐一旦确诊aHUS应立即开始依库珠单抗治疗且终身维持[1,5],也有指南主张在进行5次PE后病情无改善趋势,再转用依库珠单抗治疗[3]。指南推荐成人依库珠单抗用法及剂量:静脉滴注,900 mg/次,1次/周,连用4周后,第5周1 200 mg/次,1次/周,第6周开始1 200 mg/次,隔周1次[5]。DGKE与补体系统无直接关系,DGKE介导的aHUS使用依库珠单抗治疗可能无效[12]。依库珠单抗完全抑制补体途径后,脑膜炎球菌感染机会可能增加,指南推荐使用该药2周前接种脑膜炎球菌疫苗;未接种疫苗者,则宜在用依库珠单抗同时,给予相应的预防性抗生素治疗[5]。依库珠单抗是近年aHUS治疗的重大进展,但目前因其价格昂贵,临床使用受到一定限制。
TTP主要特征为严重的血小板减少症,MAHA和(或)不同程度的缺血性器官损伤(特别是脑、心脏和肾脏)。典型者为临床"五联征",即MAHA、血小板减少、神经精神异常、发热及肾脏损害[13,14]。
1947年Singer等[15]首次使用TTP这一术语。1966年Amorosi等[16]描述了TTP常见的5个临床特征即"五联征"。1982年Moake等[17]发现TTP患者中存在大量异常的vWF多聚体。2001年Soejima等[18]发现裂解vWF多聚体的金属蛋白酶为ADAMTS13。2002年Bianchi等[19]发现在TTP患者中大多存在ADAMTS13活性明显下降,且<10%。近年来,随着对TTP研究的深入,TTP发病机制日渐明确,主要涉及ADAMTS13基因突变或ADAMTS13抗体的产生导致其活性下降,进而形成超大vWF多聚体、血小板血栓等[6]。
临床上将TTP分为遗传性(先天性)和获得性TTP。遗传性(先天性)TTP主要是因ADAMTS13基因突变或缺乏,导致该酶活性严重下降(活性<5%),其常在感染、应激、妊娠等诱因下发病[6,20]。获得性TTP又可分为特发性与继发性TTP,特发性TTP多因体内存在ADAMTS13自身抗体(酶抑制物)导致ADAMTS13活性下降(活性<10%),其为TTP的主要临床类型;继发性TTP通常ADAMTS13活性正常或轻度下降[6,20]。
TTP临床诊断思路:当出现不明原因的MAHA、血小板减少、神经精神症状即TTP"三联征"时应高度疑诊TTP,做出初步诊断,并及时检测血浆ADAMTS13活性及其抑制物(自身抗体),同时寻找或排查HUS及其他因素导致的TMA。
经典的TTP五联征临床现已不多见,仅占20%~40%,且多为病程晚期;TTP"三联征"临床多见,占60%~80%;TTP早期可能仅表现"二联征"(血小板减少+MAHA)[14]。"五联征"的特异性相对较高;而"二/三联征"敏感性更好,有助于早期警觉、诊断[13,14]。
(1)TMA常规检查:血小板减少至<150×109/L或下降幅度>25%,凝血功能正常;血红蛋白极度下降,网织红细胞升高;结合珠蛋白降低、间接胆红素升高、乳酸脱氢酶升高、外周血破碎红细胞>1%;Coombs试验阴性[14]。(2)血浆ADAMTS13活性及抑制物(自身抗体)检测:血浆中ADAMTS13活性显著下降(<10%)。此外,在遗传性TTP常可检测到ADAMTS13等位基因的突变[21],在特发性TTP中常检测到ADAMTS13自身抗体(多为IgG,IgM、IgA少见),但也有一部分患者检测不到ADAMTS13自身抗体,机制尚不清楚;继发性TTP患者较少发生ADAMTS13活性下降或缺乏[6,20]。血浆ADAMTS13活性及抑制物(自身抗体)检测是TTP十分重要的生物学检测手段,应积极创造条件送检[6,20]。另有学者建立了一种PLASMIC评分诊断模型,在无ADAMTS13检测结果时可辅助诊断TTP[22]。最近研究还发现在TTP急性期,ADAMTS13与其自身抗体结合的构象位点是开放的,而在缓解期其构象位点是关闭的。检测ADAMTS13构象的变化,可助于TTP的诊断与预后判断[23]。
PE为TTP首选的治疗方法。PE被用于治疗TTP后,患者生存率从<10%提高到80%~90%[24]。日本指南[20]建议初始血浆的置换量为1.5×血浆容量(PVE),1次/d,以后为1.0×PVE,1次/d,直至症状缓解,血小板计数和LDH恢复正常。我国血液病专家共识[13,14]建议置换液采用新鲜血浆或新鲜冰冻血浆,置换量推荐为每次2 000 ml(或为40~60 ml/kg),1次/d,直至症状缓解,血小板计数和LDH恢复正常,以后可逐渐延长治疗间隔。继发性TTP患者PE治疗常无效,其治疗重点是处理原发疾病[13]。
PE是血液净化(广义)的一种,同属体外循环治疗技术,必须做抗凝处理,而伴有高危出血倾向的患者是使用抗凝剂的禁忌。高危出血患者的血液净化抗凝问题是制约血液净化临床应用的"瓶颈"。有学者针对该难题致力研发一种具有特殊抗凝性能的血液净化膜滤器,使其在血液净化凝血发生的关键部位,发挥局部的高效抗凝作用[25]。这种全新的血液净化抗凝理念与模式,可望应用于TTP、aHUS伴有高危出血倾向的患者。
糖皮质激素可稳定血小板、内皮细胞膜,减轻血管内皮损伤;抑制巨噬细胞活性,减少其对血小板、红细胞破坏;抑制淋巴细胞,减少自身抗体的产生。ADAMTS13抗体阳性的获得性TTP常需在PE的同时予以糖皮质激素及其他免疫抑制剂治疗,并可取得较好疗效。关于糖皮质激素的具体用法,文献报道[26]在TTP急性期使用大剂量甲泼尼龙(10 mg·kg-1·d-1,静脉滴注3 d后改为2~2.5 mg·kg-1·d-1)比常规剂量(1 mg·kg-1·d-1)治疗效果好,但目前尚未有临床随机对照试验证明PE联合糖皮质激素治疗效果优于单独PE治疗[20,27]。我国2012年TTP专家共识[14]建议对发作期TTP辅助使用甲泼尼龙(200 mg/d)或地塞米松(10~15 mg/d)静脉滴注,3~5 d后过渡至泼尼松(1 mg·kg-1·d-1),直至病情缓解。需指出的是,对ADAMTS13基因突变所致的遗传性TTP,糖皮质激素治疗常无效,临床上应从严把握其适应证。
利妥昔单抗为B细胞表面抗原CD20的单克隆抗体,可直接诱导B细胞凋亡,减少抗体产生。利妥昔单抗联合PE治疗可加快获得性TTP患者ADAMTS13活性的恢复以及ADAMTS13抗体的清除,对难治和复发性特发性TTP有一定疗效,可减少复发。多个指南建议利妥昔单抗的推荐剂量为每周375 mg/m2,连续应用4周[20,27]。
长春新碱(VCR)能改变血小板膜上糖蛋白受体,阻止vWF多聚体附着,从而抑制血小板聚集,主要用于难治性TTP的辅助治疗、PE治疗失败或反复复发者。日本指南推荐用量为1 mg/d静脉滴注。环磷酰胺、环孢素等药物作用不确切、存在争议[14]。
(1)抗血小板治疗:TTP是由血小板聚集而引发,理论上可辅助抗血小板治疗,但临床效果不确切,仅在病情趋稳后谨慎使用(阿司匹林、双嘧达莫),以减少复发[27]。(2)输注血小板:输注血小板可能会加重血小板聚集和微血栓形成,加重病情,适得其反,指南提出只有在出现危及生命的出血或行有创操作时,才慎重考虑输注血小板[28]。(3)贫血治疗:贫血严重时可输去白细胞的洗涤红细胞,一般不需要补充铁剂[14]。(4)脾切除/脾栓塞:脾切除/脾栓塞可减少TTP复发,但很少使用[27]。(5)免疫球蛋白治疗:可尝试作为难治性TTP出现新的神经症状时的辅助治疗[29]。(6)靶向治疗:重组ADAMTS13剪切酶和抗vWF纳米抗体(caplacizumab)研究近期已进入临床试验,有望分别应用于遗传性TTP和获得性TTP的治疗[30,31]。
TMA近年研究进展主要集中在以下几个方面:(1)多数aHUS患者存在补体替代途径基因突变或存在CFH自身抗体,导致补体替代途径过度活化而引发TMA;(2)TTP患者大多存在ADAMTS13基因突变或ADAMTS13自身抗体,造成vWF多聚体不能有效降解,导致血小板血栓形成,而引发TMA;(3)生物治疗已成功应用于临床,如依库珠单抗治疗aHUS,利妥昔单抗应用于ADAMTS13自身抗体所致TTP。随着对TMA认识的深入,各种继发性TMA的发病明显增加,需要给予特别关注。目前,由我国主导或参与的TMA临床指南及规范尚十分缺乏,期待我国多学科一道努力,加强循证医学的临床研究,快速提高我国TMA临床诊疗水平。

























