
探讨1个中国不典型神经轴索营养不良家系临床表现、影像学特点及基因突变情况。
收集2016年7月至河南省人民医院就诊的1个神经轴索营养不良家系,分析该家系中患者的临床表现及影像学特点,同时对先证者进行全外显子基因检测,捕获致病位点,对致病位点进行一代测序验证,并对家系中其他4名成员、100名家系外正常个体进行突变位点验证。
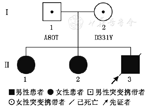
该家系中先证者(Ⅱ3)发病年龄17岁,临床主要表现为走路不稳,智力障碍、言语不清、肌张力障碍、癫痫、自主神经功能障碍,影像学表现为小脑萎缩、基底节区异常铁沉积。基因检测发现PLA2G6基因第3号外显子发生p.A80T,第7号外显子发生p.D331Y复合杂合突变,先证者两位姐姐(Ⅱ1,Ⅱ2)亦存在相同突变,先证者父亲携带p.A80T突变,母亲携带p.D331Y突变。先证者大姐(Ⅱ1)10岁左右发病,症状与先证者相似;先证者二姐(Ⅱ2)24岁开始出现步态异常;先证者两位姐姐(Ⅱ1,Ⅱ2)影像学均表现为小脑萎缩。
我们在中国人群中发现了PLA2G6基因复合杂合突变相关的不典型神经轴索营养不良家系,即p.A80T与p.D331Y突变,此突变很可能与该家系的发病相关。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
磷脂酶A2相关神经退行性疾病(PLAN)是一类以基底节、黑质及其邻近区域铁沉积以及小脑萎缩为特征的常染色体隐性遗传性疾病[1],是脑组织铁沉积性变性疾病(neurodegeneration with brain iron accumulation,NBIA)的一种亚型,即NBIA2型[2,3]。其发病率为1/1 000 000[1],致病基因为磷脂酶A2G6(PLA2G6)基因,临床可以分为三型:典型的神经轴索营养不良(infantile neuroaxonal dystrophy,INAD)、不典型的神经轴索营养不良以及帕金森病14型(PARK14)[3]。典型的INAD较常见,不典型NAD和PARK14相对较罕见[1]。INAD与不典型NAD相似,其病理均以中枢和周围神经存在轴索"球形体"为特征[4]。现我们报道由磷脂酶A2G6(PLA2G6)基因p.A80T和p.D331Y复合杂合突变所致的1个不典型神经轴索营养不良家系,并对该家系患者的临床表现及影像学特点进行分析。
收集2016年7月6日就诊于郑州大学人民医院神经内科、临床诊断为不典型神经轴索营养不良的先证者及其家系(图1)。诊断标准参照1999年临床诊断标准[5]及PLA2G6基因诊断[6]。同时纳入100名家系外正常个体进行对照。
本研究为回顾性研究,通过郑州大学人民医院伦理委员会批准,所有受试者均签署知情同意书。
详细询问并收集家系先证者(Ⅱ3)及其家系成员中有症状患者的临床资料。对先证者及其两位姐姐(Ⅱ1,Ⅱ2)行生化检查包括:血常规、肝肾功能、甲胎蛋白、红细胞沉降率、C反应蛋白、叶酸、维生素B12等指标检测。收集该家系中先证者及其两位姐姐的外院头颅磁共振(MRI)影像资料。
抽取该家系所有成员(Ⅰ1、Ⅰ2、Ⅱ1、Ⅱ2、Ⅱ3)及100名家系外正常个体外周血5 ml,经乙二胺四乙酸二钾抗凝,用标准方法提取DNA。采用全外显子测序技术对先证者(Ⅱ3)DNA进行测序,捕获目标突变位点(PLA2G6基因p.A80T和p.D331Y),并应用聚合酶链反应(PCR)对该家系的所有成员的DNA进行目标位点一代测序验证。PLA2G63号外显子引物序列:正义:5′CGTTAGTG- AGCGACCATGC3′;反义:5′TGATTCCAGCAGGG- ATGTG3′;7号外显子引物序列:正义:5′AGCTG- ACGATAGGAGGGAGAC3′;反义: 5′CTCAGAGCA- GAAGTGGCAGTG3′。PCR反应体系50 μl,包括:预混合酶25 μl,上、下游引物各4 μl(10 μmol/L),目的DNA 5 μl,去离子水13 μl。PCR反应条件:94 ℃预变性5 min,94 ℃变性30 s,退火30 s,72 ℃延伸1 min,共35个循环,72 ℃延伸10 min。PCR扩增产物纯化后送北京华大基因公司进行DNA测序。
该家系种先证者(Ⅱ3),17岁时无明显诱因出现走路向后摔倒,2~3个月后逐渐出现不能自行走路(协调不好);至当地医院行头颅MRI检查,回示:基底节区异常铁沉积,小脑萎缩(图2)。18岁时逐渐出现全身肌张力增高,言语不清,生活不能自理,智力下降;上述症状进行性加重;至我院就诊,行体格检查示:面部表情呆,记忆力下降,思维迟钝,情感淡漠,智能下降,言语不清,颅神经检查正常,双上肢肌力5级,肌强直,腱反射亢进,Hoffmann征阳性,双下肢肌力为5级,肌张力增高,膝反射亢进,踝反射亢进,踝阵挛阳性,双侧Babinski征阳性,Pussep征阳性,Chaddock征阳性。四肢深浅感觉不配合,共济运动不配合。给予对症支持治疗,效果不佳。电话随访,患者19岁时出现频发癫痫,进食困难,伴大小便失禁,伴双上肢冰凉、汗多,进行性加重,20岁去世。
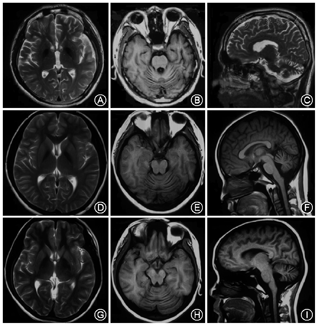
先证者大姐(Ⅱ1),10岁左右开始出现走路身体摇晃,15岁时至当地医院行头颅MRI检查,结果回示:小脑萎缩(图2);23岁时出现走路易跌倒,不协调,上述症状进行性加重;24岁开始出现智力下降、言语不清、吞咽困难、大小便失禁。2018年1月25日随访,患者25岁,查体与先证者相似。
先证者二姐(Ⅱ3),22岁开始走路姿势稍异常,自小视力差,余无特殊;2018年1月25日随访时24岁,查体视力差,共济稍差,余无特殊。其于14岁时(无症状)于当地行头颅MRI检查结果回示:小脑萎缩(图2)。
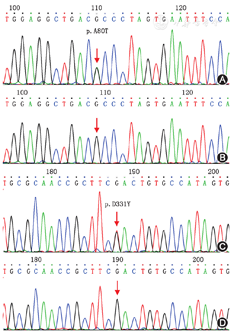
基因检测结果显示,先证者Ⅱ3存在PLA2G6基因第3号外显子发生g.46476G>A碱基突变,使编码PLA2G6蛋白的第80个位点密码子由GCC变为ACC,氨基酸由丙氨酸替换为苏氨酸,发生Ala80Thr(A80T)突变;同时PLA2G6基因第7号外显子发生g.59184G>T碱基突变,使编码PLA2G6蛋白的第331个位点密码子由GAC变为TAC,氨基酸由天冬氨酸替换为酪氨酸,发生Asp331Tyr(D331Y)突变。该突变在先证者家系中得到验证,先证者父亲Ⅰ1携带PLA2G6基因p.A80T突变,先证者母亲Ⅰ2携带PLA2G6基因p.D331Y突变,先证者两位姐姐(Ⅱ1,Ⅱ2)存在与先证者相同突变。100名家系外正常个体均未发现该位点突变(图3)。
先证者及其两位姐姐血常规、肝肾功能、甲胎蛋白、红细胞沉降率、C反应蛋白、叶酸、维生素B12等抽血指标检测均正常。
本研究报道了1个由PLA2G6基因突变所致不典型神经轴索营养不良家系的临床及影像学特征。典型的INAD患儿通常于6个月至3岁起病,临床以进行性运动障碍、智力低下,躯干性肌张力障碍、锥体束征阳性以及视力障碍为特征,部分患者晚期合并有癫痫发作[7];病情进展较快,患儿多于10岁前去世[8]。与INAD患者相比,不典型NAD有较大的表型变异,发病年龄较晚,多于4岁左右起病[9],有的也可晚至20岁[3],病情进展较慢,但其临床症状体征与INAD相似,可伴有不同形式的步态不稳、共济失调、社交障碍、言语障碍以及自闭症特征[4,7]。INADA与不典型NAD影像学均可表现为小脑萎缩伴或不伴有基节区异常铁沉积[10]。本研究纳入的家系特点主要为:发病年龄从17岁、10岁、22岁不等,临床表现早期以步态不稳,各种形式的共济失调为特征,后期伴有智能下降、言语不清、肌张力障碍、锥体束征阳性,三位患者影像学均表现为小脑萎缩,先证者影像学伴有基底节区异常铁沉积,另两位患者不伴有铁沉积。与文献报道的不典型神经轴索营养不良临床特征相一致。但患者(Ⅱ1、Ⅱ3)晚期伴有自主神经功能障碍,考虑为疾病晚期机体全面退行性变,累及自主神经所致。
根据人类基因组(HGMD)数据库,PLA2G6基因A80T和D331Y突变均被报道过为PLAN致病性突变。其中A80T突变报道过1次,临床诊断为NBIA,影像学伴有基底节区异常铁沉积但没有具体描述其临床表现,不清楚其具体临床分型[11],D331Y突变报道过多次与PARK14相关[12,13,14]。本研究报道的家系为A80T及D331Y复合杂合突变,3例患者中仅有1例影像学表现为基底节区异常铁沉积,3例均符合不典型NAD报道,与文献报道的A80T家系及D331Y家系表型不同,这可能与种族不同、其他基因的相互作用以及环境因素的影响有关。
PLA2G6基因由17个外显子组成,编码产生由806个氨基酸组成的胞质内非Ca2+依赖性第Ⅵ组磷脂酶A2(iPLA2)蛋白[15]。该蛋白在细胞膜结构的生成和稳定上起关键作用。致病性突变遍布于从N端到C端各个区域,绝大部分突变的机制可能为干扰细胞膜稳态和蛋白质降解过程,引起神经轴索变性[6]。至于p.A80T与p.D331Y具体致病机制尚不清楚,有待于进一步研究。
诊断此病时需临床与基因检测相结合,注意与以下几种具有类似MRI表现的疾病相鉴别。一氧化碳中毒性脑病、甲醇中毒性脑病等代谢中毒性疾病,均可表现为双侧苍白球对称性病变,但均表现为双侧苍白球T2WI信号增高,没有T2WI低信号改变,结合相关病史即可明确诊断;进行性核上性麻痹有特征性的核上性凝视麻痹,有智力障碍和步态异常,多为中老年患者发病,MRI显示中脑萎缩。肝豆状核变性为遗传性铜代谢障碍引起的肝硬化和脑变性疾病,患者有肝硬化病史,血清铜蓝蛋白显著降低,可见角膜色素环K-F环,双侧豆状核T2WI呈高信号改变;少年型亨廷顿病以弥漫性脑萎缩为主,头颅MRI常可见尾状核头部和壳核萎缩[16]。
我们在中国1个临床表现为不典型神经轴索营养不良家系中发现发现了PLA2G6基因的2个突变位点,即PLA2G6基因第3号外显子A80T突变以及第7号外显子D331Y突变,该复合杂合突变很可能与该家系患者的发病相关。本研究在一定程度上丰富了国内PLA2G6基因数据库。
所有作者均声明不存在利益冲突

























