
特发性炎性肌病(IIM)是一组获得性免疫性肌病,其主要包括多发性肌炎、皮肌炎、无肌病性皮肌炎、散发性包涵体肌炎(sIBM)和免疫介导的坏死性肌病,以及有些特殊类型的抗合成酶抗体综合征、抗信号识别颗粒抗体阳性坏死性肌病(NM)、抗3-羟基-3-甲基戊二酰辅酶A还原酶抗体阳性NM。这些不同类型IIM的诊断主要依赖临床表现、抗体检测和肌肉病理技术。不同类型的IIM,其临床表现不同,之间还存在着相互重叠的表现。文中就IIM类型的演变、肌炎的主要抗体、肌肉病理特点、各类型表现以及治疗进行系统描述。除外sIBM,其他类型的IIM患者在经过早期诊断和及时正确足量的药物治疗后,多数均有好的临床结局。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
特发性炎性肌病(idiopathic inflammatory myopathies,IIM)是一类免疫介导的骨骼肌非化脓性炎性疾病,也是自身免疫性疾病的一大类型[1,2,3]。早期的IIM特指多发性肌炎(polymyositis,PM)和皮肌炎(dermatomyositis)。随着肌肉病理的广泛开展以及越来越多相关抗体的发现和应用,IIM的类型不断增加,诊断标准也在不断地更新。尽管如此,对IIM的诊断和治疗仍遵循以临床为主、病理为关键、相关抗体为辅的原则。
由于IIM是一个涉及多系统受累的疾病,因此该类患者经常在不同的临床学科就诊,如神经内科、风湿科、皮肤科等。不同学科的专家对IIM的诊治和研究会得出不同的结果和观点,尤其IIM的分类又较多。皮肌炎和PM是古老的疾病,早在100多年前就被人们所认识,Wagner分别在1863年和1887年就描述了皮肌炎和PM的病例[2,4,5]。后来,Unverricht又在1887年和1891年分别对PM和皮肌炎进行系统地描述,提出了这两个疾病的概念;Gowers在1899年还发表了PM的专题讲座,使人们比较系统地认识了皮肌炎和PM[2,6,7,8]。从此,PM和皮肌炎作为IIM的两个类型一直被广泛应用于临床和相关研究。1975年,Bohan和Peter发表了著名的IIM分类:(1)原发性PM;(2)原发性皮肌炎;(3)合并血管炎的儿童PM或皮肌炎;(4)合并肿瘤PM或皮肌炎;(5)合并其他结缔组织病的PM或皮肌炎[9]。该分类一直被临床沿用至今。
早在1965年,Adams等[10]首次描述了有包涵体的肌病病例;Yunis和Samaha[11]在1971年正式提出包涵体肌炎(inclusion body myositis,IBM)的概念。次年,Carpenter等[12]报道了IBM患者的临床与病理特点后得到同行认可;至此,IBM才被认可为一个独立的新病种。1991年,Massa等[13]报道了1个遗传的IBM家系,之后该类家族遗传病例报道逐渐增多,并确认了该病的存在并冠名为遗传性包涵体肌病(heredity inclusion body myopathies,hIBM)。为了与hIBM相区别,人们又将获得性IBM称为散发性IBM(sporadic inclusion body myosistis,sIBM)。同年,Dalakas[14]将IIM又简单地分为PM、皮肌炎和sIBM 3个类型并提出了诊断标准和鉴别诊断的条件。Griggs等[15]在1995年提出了针对sIBM更加具体的诊断标准并沿用至今。
同年,Krain[16]首次报道了6例具有典型的皮肌炎皮肤损害而没有肌炎表现的病例,Pearson[17]在1979年将其命名为"无肌病性皮肌炎(amyopathic dermatomyositis,ADM)",Euwer和Sontheimer[18]在1991年提出了ADM的定义,即"无明显肌肉损害证据但又具备典型的皮肌炎皮损表现";他们又在1993年提出了ADM的诊断标准[19];Sontheimer[20]在1999年又修改了ADM的诊断标准并强调,ADM作为皮肌炎的一个临床亚型,属于自身免疫性疾病或副肿瘤综合征的表现之一。故将IIM再分为6个类型:(1)原发性PM;(2)原发性皮肌炎;(3)儿童PM或皮肌炎;(4)肿瘤相关的PM或皮肌炎;(5)伴结缔组织病的PM或皮肌炎;(6)ADM。
1969年,Smith[21]报道了1例结肠癌和1例乳腺癌患者出现四肢近端无力、肌肉疼痛,2例患者的肌肉病理均发现有明显的肌纤维坏死,但1例有极少量淋巴细胞浸润,1例没有炎性细胞,经肿瘤切除后,病情明显改善,因而首次提出了坏死性肌病(necrotizing myopathy,NM)的概念。1991年,Emslie-Smith和Engel[22]进一步研究发现,NM的病理特点更支持其独立性,后来的许多研究不仅提示NM与恶性肿瘤有关,还可以是单纯的免疫介导所致。不过,NM一直未被列入IIM的分类中。直到2004年欧洲神经肌肉病中心对IIM又提出了新的分类,即PM、皮肌炎、sIBM、非特异性肌炎(non-specific myositis,NSM)和免疫介导的NM(immune-mediated necrotizing myopathy,IMNM)[23]。主要依据为肌肉的病理特点。但在该分类中,难以界定的是NSM。而后,Dalakas[24]在2015年从临床、抗体和病理角度,又将IIM分为PM、皮肌炎、sIBM、IMNM和重叠性肌炎(overlap myositis,OM)5个类型。
随着临床免疫学研究的进展,特别是许多与IIM有关的肌炎特异性抗体(myositis-specific autoantibodies,MSAs)被相继发现后,又有许多新的概念和名词提出。如Nishikai和Reichlin[25]在1980年发现抗组氨酰tRNA合成酶(Jo-1)抗体及之后发现的更多抗合成酶抗体,而提出的抗合成酶抗体综合征(antisynthetase syndrome,ASS);Reeves等[26]于1986年在PM患者血清中发现抗信号识别颗粒(signal recognition particle,SRP)抗体,而后发现抗SRP抗体主要存在于NM患者,因此称为抗SRP抗体阳性NM;Sato等[27]在2005年发现抗黑色素瘤分化相关蛋白5(melanoma differentiation-associated gene 5,MDA5)抗体与皮肌炎关系非常密切,而命名为抗MDA5抗体阳性皮肌炎;Christopher-Stine等[28]于2010年在NM患者血清中发现抗3-羟基-3-甲基戊二酰辅酶A还原酶(3-hydroxy-3-methylglutaryl-coenzyme A reductase,HMGCR)抗体而命名为抗HMGCR抗体NM等,这些肌炎的特异性相关抗体均存在于IIM中,有的甚至以IIM为首发表现。因此,IIM的分类不得不将各种MSAs考虑在内。为此,2018年欧洲抗风湿联盟/美国风湿学会又提出了IIM的分类及诊断标准,即PM(包含IMNM)、sIBM、ADM、皮肌炎、幼年型PM和幼年型皮肌炎[29]。这种分类综合临床表现、抗体和病理权重给予评分,以判断IIM诊断的强度级别,但是这种分类的层次欠合理,如将PM与IMNM混为一型,皮肌炎和幼年皮肌炎(JDM)分为两个独立的类型,这并不利于临床实践的应用。Selva-O′Callaghan等[30]在2018年提出了成人IIM的分类标准,即皮肌炎、IMNM、sIBM、OM和PM;其中特别提出OM包括ASS。该分类把ASS列入IIM之中,特别提及应注意鉴别ASS与其他结缔组织病,甚至恶性肿瘤相关的肌炎。
总之,在IIM分类的演变过程中,不同学科的专家从自身的角度,把IIM分为不同的类型,各有利弊,还不能完全统一。但不管如何分类,目前的IIM应该主要包括PM、皮肌炎、ADM、sIBM和IMNM。另外,也应特别注意与MSAs有关的综合征和疾病,如ASS、抗SRP抗体阳性NM或肌病、抗HMGCR抗体阳性NM和抗MDA5抗体阳性皮肌炎等[1,31,32,33,34,35,36,37]。这些亚型在许多临床学科中均可见到,且必须通过临床表现、相关抗体、神经电生理、影像学及肌肉活体组织检查(活检)方可明确诊断和鉴别诊断。
许多在IIM患者中检测到的抗体可分为两大类,肌炎相关性抗体(myositis-associated autoantibodies,MAAs)和MSAs。MAAs是指这种抗体不仅出现于IIM患者,还可以出现在其他免疫性疾病患者中,这类抗体主要包括抗PM-系统性硬化症(PM-Scl)抗体、抗Ku抗体、抗核糖体核蛋白(RNP)抗体、抗SSA(Ro)52 kDa抗体、抗SSa(Ro)60 kDa抗体、抗SSB(La)抗体和抗U1snRNP等。MSAs是指仅主要出现在IIM患者中,包括抗氨基酰rRNA合成酶(aminoacyl-tRNA synthetase,ARS)系列抗体、抗SRP抗体、抗HMGCR抗体、抗Mi-2抗体、抗MDA5抗体(又称抗临床无肌病性皮肌炎-140抗体)、抗转录调节因子1γ(TIF1γ)抗体(又称抗p155/p140抗体)、抗核基质蛋白2(NXP-2)抗体[又称抗MJ抗体或抗DNA错配修复蛋白(PMS1/2)抗体]、抗小泛素样修饰酶(SAE)抗体、抗胞质5核苷酸酶1A(cytosolic 5′nucleotidase,cN1A)抗体等。通过检测这些MSAs,有助于IIM的分类、治疗和预后判断[34,35,36,37,38]。
目前共有8种抗ARS抗体,即抗Jo-1抗体、抗苏氨酸tRNA合成酶(PL-7)抗体、抗丙氨酸tRNA合成酶(PL-12)抗体、抗甘氨酰tRNA合成酶(EJ)抗体、抗异亮氨酸tRNA合成酶(OJ)抗体、抗天冬酰胺基tRNA合成酶(KS)抗体、抗苯基丙氨酰tRNA合成酶(Zo)抗体和抗酪氨酰tRNA合成酶(Tyr)抗体。抗体ARS抗体阳性的肌炎患者一般被诊断为ASS,抗ARS系列抗体的阳性率差别较大,抗Jo-1抗体阳性率最高,其他7种抗ARS抗体阳性率均较低(表1)[39]。因此,ASS也称为抗Jo-1抗体综合征。ASS是一组多系统受损的症候群,包括肌炎、间性肺病变(interstital lung disease,ILD)、关节炎、雷诺现象、技工手、发热及皮疹等,但其发生率不同,如肌炎发生率为82%~100%、ILD 23%~79%、多关节炎50%、雷诺现象60%~93%、发热80%、技工手70%、干燥性角膜结膜炎59%等。抗ARS抗体主要出现在PM和皮肌炎中(20%~29%),其次为肿瘤相关肌炎(13%);甚至有2%的sIBM也出现该抗体[34,35,36,37,40,41,42,43,44]。所以,临床上考虑为IIM者,应该检测抗ARS抗体;特别是出现不明原因的ILD时,应检测抗ARS抗体,以及时鉴别诊断和治疗。ASS的诊断条件是:(1)血清抗ARS抗体必须为阳性;(2)至少有1个或多个以下表现:明确的肌炎、肯定的ILD、关节炎、技工手、雷诺现象及不明原因的发热[39]。
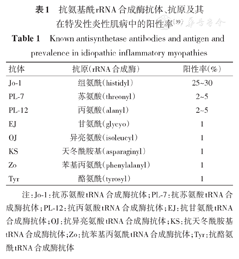
抗氨基酰rRNA合成酶抗体、抗原及其在特发性炎性肌病中的阳性率[39]
Known antisynthetase antibodies and antigen and prevalence in idiopathic inflammatory myopathies
抗氨基酰rRNA合成酶抗体、抗原及其在特发性炎性肌病中的阳性率[39]
Known antisynthetase antibodies and antigen and prevalence in idiopathic inflammatory myopathies
| 抗体 | 抗原(rRNA合成酶) | 阳性率(%) |
|---|---|---|
| Jo-1 | 组氨酰(histidyl) | 25~30 |
| PL-7 | 苏氨酸(threonyl) | 2~5 |
| PL-12 | 丙氨酸(alanyl) | 2~5 |
| EJ | 甘氨酰(glycyo) | 1 |
| OJ | 异亮氨酸(isoleucyl) | 1 |
| KS | 天冬酰胺基(asparaginyl) | 1 |
| Zo | 苯基丙氨酰(phenylalanyl) | 1 |
| Tyr | 酪氨酰(tyrosyl) | 1 |
注:Jo-1:抗苏氨酸tRNA合成酶抗体;PL-7:抗苏氨酸tRNA合成酶抗体;PL-12:抗丙氨酸tRNA合成酶抗体;EJ:抗甘氨酰tRNA合成酶抗体;OJ:抗异亮氨酸tRNA合成酶抗体;KS:抗天冬酰胺基tRNA合成酶抗体;Zo:抗苯基丙氨酰tRNA合成酶抗体;Tyr:抗酪氨酰tRNA合成酶抗体
SRP是一种广泛存在于细胞质中的核糖核酸蛋白复合体,其开始先聚集在细胞核内,稳定后被运输到胞质内。在受到物理、化学或生物因素的影响下,SRP发生免疫原性改变,被自体误认为是异已抗原而遭到攻击;SRP分子被切割成多个有趋化活性的抗原多肽片段而成为内源性抗原;再通过巨噬细胞、CD4+T细胞、CD8+T细胞和树突细胞的抗原呈递作用,激活的T淋巴细胞和B淋巴细胞而启动免疫应答机制,分泌抗SRP抗体和细胞因子,导致毛细血管出现C5b-9沉积和肌纤维发生坏变而引发NM[38,45]。早在1986年[26],就有学者发现PM患者中存在抗SRP抗体,后来发现在其他IIM患者中也存在该抗体。直到2002年Miller等在分析一组IIM患者的肌肉病理特点后,首次提出抗SRP抗体肌病的概念,认为其是免疫性NM的常见类型,故称为抗SRP抗体阳性NM;在皮肌炎、PM等其他IIM患者中检测到抗SRP抗体阳性者则称为抗SRP抗体阳性肌病[45]。SRP抗体在不同类型的IIM患者中阳性率差异较大,如在IIM患者中的总阳性率为4%~5%,在成人PM/皮肌炎患者中的阳性率为8%~13%,在JDM患者中阳性率低至2%,而在NAM患者中阳性率高达15%。一般认为,抗SRP抗体阳性NM患者的肌肉受损较重,如颈肌无力、吞咽困难、呼吸困难,甚至较早出现肌肉萎缩,血肌酸激酶水平显著升高,且治疗效果不佳,只有少数治疗效果良好的病例[34,35,38,45,46,47,48,49]。
抗HMGCR抗体是在NM患者的血液中发现的[28],而后许多研究发现抗HMGCR抗体与他汀类药物的使用有关;临床研究报告在抗HMGCR抗体阳性的NM患者中,有66.66%(30/45)有他汀类药物的服用史,如限于50岁以上患者进行统计,则比例更是高达92.31%(24/26),提示抗HMGCR抗体与他汀类药物有密切关系[28]。不过,也有临床研究结果并非如此,如另一项研究检测了405例IIM患者,只有5.4%(22例)抗HMGCR抗体呈阳性,而其中只有3例服用过他汀类药物[50];还有报道称PM患者的抗HMGCR抗体阳性率为18.4%,皮肌炎为15.2%,甚至幼年炎性肌病患者抗HMGCR抗体的阳性率高达15%。这又说明有不少抗HMGCR抗体阳性的NM患者与他汀类药物的使用无关;还有报道长期服用他汀类药物又不出现IIM的患者,均检测不到抗HMGCR抗体,提示仅服用他汀类药物并不能诱导产生抗HMGCR抗体。抗HMGCR抗体阳性的IMNM患者,其主要的临床表现为急性或亚急性发病的对称性近端肌无力,部分出现肌肉疼痛、吞咽困难、关节痛、皮疹、雷诺现象等;血清肌酸激酶升高达正常值上限的10倍以上;肌电图提示为肌源性受损;肌肉MRI显示肌肉水肿明显并伴有筋膜水肿,后期还可显示肌肉萎缩及结缔组织增生和脂肪化;有近1/4的患者生活不能自理[51,52,53]。因此,不论IIM患者是否服用他汀药物,都应该检测抗HMGCR抗体,以协助判断IIM的类型。抗HMGCR抗体阳性的IMNM应该分为"他汀类药物相关的抗HMGCR抗体阳性的IMNM"和"他汀类药物不相关的抗HMGCR抗体阳性的IMNM",其诊断标准为:(1)临床特点为亚急性或隐性发病,对称性肢体无力,近端重于远端,颈屈肌明显无力;(2)血清肌酸激酶明显升高;(3)血清抗HMGCR抗体阳性;(4)肌电图呈肌源性受损;(5)MRI提示肌肉弥漫性或局灶性高信号;(6)肌肉病理提示为典型的NM病理改变。他汀类药物相关的抗HMGCR抗体阳性的IMNM和他汀类药物不相关的抗HMGCR抗体阳性的IMNM的诊断标准区别在于前者可询问出长期应用他汀类药物,后者没应用他汀类药物史[23,35,52,53,54]。
也称为抗临床无肌病性皮肌炎(CADM)-140抗体,最初是在CADM合并ILD患者的血清中被发现[27]。MDA5也称为干拢素诱导的解旋酶C结构域(interferon induced with helicadse C domain 1,IFIH1),主要参与免疫性防御反应;当皮肤或肺上皮细胞感染病毒后,导致MDA5蛋白水解的片段和病毒核糖核酸与MDA5复合体的释放,进而对被感染的细胞产生细胞毒性作用,也导致抗MDA5抗体的产生,后者引起自身抗体依赖的细胞毒性作用而产生自我损害[55,56]。抗MDA5抗体被认为是皮肌炎的特异性抗体,在皮肌炎患者中的阳性率为20%~50%,DADM为50%~73%。抗MDA5抗体与并发ILD有明显相关性,抗MDA5抗体在皮肌炎患者并发ILD中的阳性率高达90%~95%;超过半数的抗MDA5抗体阳性CADM患者出现ILD。抗MDA5抗体阳性的ADM患者皮肤溃烂、口腔溃疡、关节炎等明显突出。抗MDA5抗体阳性的患者容易出现快速进展的肺间质病变(RPILD)风险,病死率高达50%。总之,抗MDA5抗体与皮肌炎/CADM和ILD有明确的相关性,通过检测抗MDA5抗体不仅能判断皮肌炎/CADM是否可能合并ILD,也可预测是否发生RPILD;动态观察该抗体变化有助于评价皮肌炎/CADM与ILD的预后[35,55,56,57,58]。
该抗体最早在1976年被Reichlin和Mattioli[59]在1例60岁女性皮肌炎患者的血清中发现,后来确定该抗体是皮肌炎的特异性抗体[60],其在成年皮肌炎患者中的阳性率为11%~59%,在JDM中为4%~10%。抗Mi-2抗体与皮肌炎的特殊皮疹有关,常出现Gotton丘疹、Heliotrope皮疹、V征和披肩征。一般来讲,抗Mi-2抗体阳性肌炎患者的病情预后较好,如肌无力较轻、并发ILD的风险低、对免疫抑制治疗反应良好[35,61,62,63]。
也称抗p155/p140抗体,其靶抗原为TIF1家族蛋白,包括抗TIF1α、TIF1β和TIF1γ抗体3种。抗TIF1抗体是皮肌炎的特异性抗体,在成人PM/皮肌炎中阳性率为13%~31%,在JDM中为22%~29%。该抗体阳性的皮肌炎患者的皮肤广泛受损,表现为手掌角化过度性丘疹、银屑样病变和红白斑块。因TIF1蛋白在肿瘤的发生中起关键作用,所以抗TIF1抗体最常见于肿瘤相关性皮肌炎患者。如果患者血清同时出现抗TIF1α和TIF1γ抗体,则提示出现肿瘤的风险明显增高。因此,皮肌炎患者的血清抗TIF1γ阳性,应积极排查肿瘤,以及早处理[35,63,64,65]。
又称抗MJ抗体或抗DNA错配修复蛋白(PMS1/2)抗体,该抗体是Oddis等于1997年在幼年肌炎患者中发现的一种新抗体,而后证明其靶抗原为NXP-2,因此将抗MJ抗体改称为抗NXP-2抗体[35]。该抗体主要出现于幼年患者,在JDM的阳性率为23%~25%,成人肌炎阳性率为1%~17%。抗NXP-2抗体阳的JDM患者临床特点为肌肉无力、萎缩和挛缩,钙质沉积显著。该抗体与恶性肿瘤密切相关,如与普通人群预期发病率比较,抗NXP-2抗体阳性者发生恶性肿瘤的风险增加3.68倍;有1/3的抗NXP-2抗体阳性IIMs患者在3年内出现肿瘤[35,63,65,66,67]。
该抗体最早由Betteridge等[68]描述,其靶抗原是小泛素样修饰物-1(SUMO-1)激活酶(SEA)异二聚体SAE1和SAE2。几个大样本临床研究表明,IIM患者的抗SAE抗体阳性率仅为4%;大部分抗SAE抗体阳性者首先出现皮损症状,82%的患者出现向阳性皮疹和Gottron丘疹;还有82%的患者出现发热、体重减轻和炎性指标升高,78%的患者出现吞咽困难,71%的患者出现轻度ILD。目前认为抗SAE抗体是皮肌炎的主要抗体,大部分抗体阳性患者首先出现皮肤病变,而后出现严重的吞咽困难;但是抗SAE抗体阳性与肿瘤没有关系;不过,抗SAE抗体阳性患者的治疗效果和预后都较好[35,68,69]。
长期以来,sIBM被认为是一种具有炎性的肌肉变性病。直到Salajegheh等在2011年发现近半数(52%,13/25)的sIBM患者血浆中可检测到一种能结合相对分子质量为43 000的肌蛋白抗体,后来证明这种蛋白为胞质5核苷酸酶1A(cytosolic 5′ nucleotidase 1A,cN1A),人们才认识到sIBM是一种免疫性疾病[35]。大样本IID临床分析提示,IBM患者的抗cN1A抗体的阳性率为33%~34%,PM为4%~5%,皮肌炎为0~4%,其他神经肌肉病为0~3%,健康人群为0。不过,该抗体还可出现在其他免疫性疾病中,如干燥综合征(36%)、系统性红斑狼疮(20%)。该抗体对IBM诊断的敏感度为49%~53%,特异度为94%~96%。故认为抗cN1A抗体是sIBM的特异性抗体,可作为sIBM的特异性血清标志物,不过仍需要更多的病例验证该抗体与临床表现、治疗效果和预后的关系[35,70,71,72,73,74]。
综上所述,MSAs对IIMs的诊断及其分型和治疗有很好的特异性与指导性,个别的MSAs对某些特殊类型的IIMs诊断也起着重要作用。因此,临床遇见疑似IID的患者,应该进行肌炎方面的抗体检测,以协助IIMs的诊断和分型。
对于IIM来讲,尽管有许多的MSAs和MAAs可协助判断IIM的各种类型,但是这些抗体仍不是最具特异性的,某种类型的IIM患者可同时出现多种抗体阳性,也有不同类型的IIM患者出现同一种抗体阳性,还可有某种类型的IIM没有检测到任何抗体阳性。此外,还有许多其他肌病与IIM有非常类似的表现,无法鉴别。因此,从临床的角度,越来越强调对疑似IIM的患者,应该做肌肉活检,以更客观地协助诊断某种IIM的类型,还可以鉴别IIM与其他非炎性肌病。值得强调的是,肌肉活检技术必须用新鲜的肌肉组织通过酶组织化学及必要的免疫组织化学技术方能正确观察到真正的肌肉病理变化,决不能按大病理技术用福尔马林固定。
不同PM在不同时期的病理改变不同。在急性期未治疗之前,苏木素-伊红(HE)染色提示有肌纤维肿胀和坏死,坏变肌纤维内出现较多的小空泡样水肿,严重者肌纤维破碎;坏变肌纤维间隙有以淋巴细胞为主的大量炎性细胞浸润;可伴有较多的核内移,坏变肌纤维或萎缩肌纤维呈角形或不规则形态为主;一般没有肌纤维肥大、分裂及增生(图1)。在慢性期,除了肌纤维坏变和淋巴细胞浸润外,可伴有明显的肌纤维肥大、增生和分裂;有的出现肌纤维严重萎缩和脱失,伴有大量结缔组织增生,尤其是脂肪细胞增生更为明显。不论是急性还是慢性期,HE染色均可观察到单核细胞浸润非坏死肌纤维是本病的病理特点(图2),不过这种现象并不容易观察到;酸性磷酸酶(ACP)染色提示坏变肌纤维及炎性细胞呈红染,提示酸性磷酸酶活性明显增强(图3);非特异性酯酶(NSE)可见坏变肌纤维及浸润的炎性细胞呈棕褐色阳性(图4);但肌纤维氧化酶,如琥珀酸脱氢酶(SDH)、还原型辅酶Ⅰ四唑氮还原酶(NADH)、细胞色素氧化酶(COX)等染色提示局灶性活性减低;ATP酶染色提示两型肌纤维均受累。免疫组织化学染色可以协助明确PM的病理特点,如大部分炎性细胞为T淋巴细胞,其中以CD8+T细胞为主,还可有吞噬细胞;最具特征的改变为肌纤维膜有主要组织相容复合体-1(major histocompatibility complex-1)的异常表达,CD8+T细胞围绕在非坏死的表达MHC-I的肌纤维周围或侵入这些肌纤维内,但是这种现象并不容易在肌肉病理中观察到,故观察不到该现象也不能排除PM[1,2,3,24,31,32,75]。
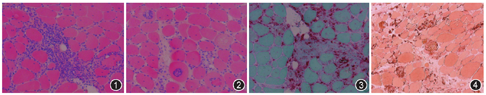
皮肌炎患者的活检分为两部分,即皮肤活检和肌肉活检。皮肤活检按常规病理技术进行,HE染色在早期可见表皮和真皮水肿坏变,表皮角化,棘层萎缩,基底细胞液化;小血管扩张,其周围有淋巴细胞浸润;后期出现胶原纤维增生,小血管壁增厚,表皮萎缩,甚至出现广泛钙化。免疫组织化学提示病灶内有免疫球蛋白及补体沉积,伴有CD4+T淋巴细胞、浆细胞和组织细胞浸润。肌肉行酶组织化学染色显示最具特征的病理改变是坏变和萎缩的肌纤维呈束周分布(图5),即坏变和萎缩肌纤维均出现在肌束的周围,而肌束中心的肌纤维一般不受累;但是这种束周分布现象并非在每例皮肌炎患者中都能观察到,出现率占半数左右。除了束周分布现象外,坏变肌纤维还表现为皱缩、空泡化,甚至破碎(图6),NADH和ACP染色提示束周分布的坏变萎缩肌纤维呈氧化酶活性增高现象[NADH为深染(图7),ACP为红染)],而COX和三磷酸腺苷酶(ATPase)染色显示为酶活性降低(浅染)[1,2,3,24,32,75,76]。
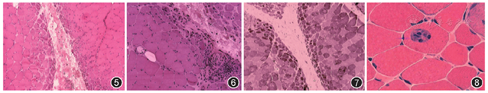
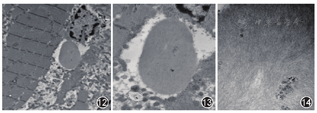
本病的诊断来源于电镜观察到坏变肌纤维内有包涵体结构而得名,因此,肌肉病理对本病的诊断是唯一的"金标准"。sIBM的病理特点为:(1)肌纤维间隙有轻至中度的炎性细胞浸润,可见单核细胞浸润非坏死肌纤维(图8),这些炎性细胞主要为CD8+T细胞;(2)坏变萎缩肌纤维出现数量不同的镶边空泡(图9),HE染色显示这些空泡内有许多嗜碱性紫蓝色颗粒(图10),改良Gomori三色(MGT)染色提示颗粒呈红染现象(图11);(3)刚果红染色观察到这些镶边空泡内的颗粒呈阳性;(4)电镜观察到肌膜下或肌核内有直径为15~18 nm的管丝状小团块结构(图12,图13,图14),有的肌膜下还有成堆的髓样体、膜样体和糖原颗粒等各异形态结构(即HE染色显示的镶边空泡内颗粒)。值得注意的是,电镜不容易观察到肌纤维内的管丝状包涵体结构,因为并非每个病变的肌纤维均存在这种包涵体,加之超薄切片,每次仅切几十个纳米(nm)的厚度,微小的包函体被切到的概率极小,所以不主张应用电镜诊断sIBM;而是结合临床特点及肌肉酶组织化学观察到较多坏变萎缩的肌纤维有镶边空泡即可考虑为本病[1,2,3,24,32,77,78,79,80]。
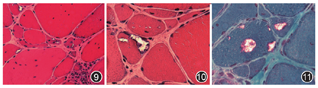
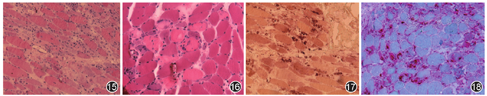
不论哪种MSAs阳性的IMNM,HE染色均可见广泛分布的大量肌纤维坏死(图15)或多发性局灶性片状肌纤维坏死(图16),且没有或极少有炎性细胞浸润;MGT、NADH、ATP酶染色提示这些坏死肌纤维的酶活性明显减低;NSE和ACP染色提示坏死肌纤维及其周围的酶活性增强,前者为深褐色(图17),后者为红色(图18)[1,2,3,24,32,33,47,48,52,81]。
虽然IIM是一组皮肤、肌肉、结缔组织和其他不同组织器官受累的炎性病变,相互之间存在许多重叠机制和表现,但是每个亚型仍存在自己的特殊表现。各个亚型实际上是指受累的相关组织表现出的相应症状和体征。
可发生在任何年龄,但以中青年发病较多,儿童极少见,伴恶性肿瘤的PM以中老年多见。可急性发病,也可亚急性发病,有不少为慢性发病;不管是急性还是亚急性发病,最终都演变为慢性过程,没有在短期内可以治愈的可能,多数病程在1年以上,有的长达十几年。PM患者的表现以双下肢近端无力为主,即起蹲困难、不能跑跳和上楼费力;随着病情进展,可累及双下肢远端、双上肢和颈肌无力;表现为不能行走,甚至不能站立,双臂上举困难,抬头费力;严重者可累及球部肌肉和呼吸肌而出现球麻痹和呼吸困难,如咀嚼无力、面部表情少、闭眼无力、苦笑面容、吞咽困难、饮水呛咳、说话不清、咳痰费力、呼吸力弱而急促,甚至口唇和四肢末端发绀。少数患者在急性或亚急性期可出现肌肉酸胀、疼痛和压痛;急性发病,病情进展快且较重者可出现横纹肌溶解,表现为酱油色尿;慢性期可出现不同程度的肌肉萎缩。一般不累及眼外肌,故无复视。单纯性PM患者不会出现其他组织器官受损的表现。PM合并结缔组织病者称为结缔组织病相关性肌炎;合并恶性肿瘤者称为肿瘤相关性肌炎。此时,患者还可表现出相关的组织器官受损的症状和体征[1,2,3,24,31,32,38,40,75,81]。
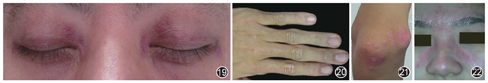
可见于任何年龄,但好发于青少年。多数缓慢起病,少数呈急性或亚急性起病,一些患者有前驱症状,如发热、咽痛、关节痛、雷诺现象等。因皮肤和肌肉受损而出现两组主要表现。皮肤受损表现常先于肌病出现数周甚至数年;也有部分患者同时出现皮肤和肌肉受损表现;但极少先出现肌肉受损后出现皮肤受损表现。皮肌炎的皮损表现非常特殊,明显有别于其他皮肤病,常见的表现有:(1)眶周水肿性紫红斑(图19;heliotrope征:指双上眼睑暗紫红色水肿性红斑):眼眶周围皮下水肿,皮肤有暗紫红斑疹。(2)Gottron征(图20,图21):指间和掌指关节背面、肘后、膝部等处出现暗紫红色的丘疹(Gottron′s papules);这些丘疹有时可融合成斑片状,伴有局部毛细血管扩张、潮红和鳞屑,则称为Gottron征。(3)暴光部位红斑:也称向阳性皮疹(helioprotic rash;图22),即在颈、肩、上胸部V区、手背、足背等暴露部位出现潮红、水肿性斑片,其界限清楚,有瘙痒感。(4)技工手:也称操作机械手(mechanic hand),即手外侧掌面皮肤出现角化、裂纹、粗糙、脱屑和色素沉着等,类似技术工人干活的手,故称"技工"手。(5)皮肤异色征:受损的皮肤出现毛细血管扩张,轻度萎缩,既伴有色素沉着,又有色素减退的现象。(6)甲周红斑:甲周毛细血管扩张,在甲周及甲小皮处出现角化、出血点、毛细血管扩张等。(7)Holster征:双侧大腿和臀部侧面出现的对称性紫红斑。(8)钙沉着:少数患者的皮肤发生钙化小结节。还有其他的皮肤损害表现,如网状青斑、黏糊损害、恶性红斑、水疱和大疱、溃疡性损害、毛发红糠疹、红皮病和瘙痒等。皮肌炎的肌肉受损表现类似于PM的症状和体征,急性或严重者出现肌肉酸胀、疼痛和压痛以及横纹肌溶解,出现酱油色尿。合并恶性肿瘤和结缔组织病者,还可表现出多系统损害,如关节肿痛,肺炎、心肌炎和心包炎等[1,2,3,24,32,38,40,75,76]。
也称无肌炎性皮肌炎,是皮肌炎的一个亚型,该病患者主要在皮肤科诊治,一般不在神经内科就诊。既然有肌肉受损的PM和肌肉与皮肤同时受损的皮肌炎,则可有仅是皮肤受损的ADM。该类型的临床表现就是上述皮肌炎皮肤受损的特殊表现,且不存在肌肉受损表现,即相关检查均没有发现有肌无力和肌酶升高,肌电图和肌肉活检均正常。Euwer和Sontheimer[19]在1993年就提出了ADM的诊断标准:(1)具有皮肌炎特征性的皮肤病变;(2)皮肤病理与皮肌炎一致;(3)皮肤病变出现后2年内没有近端肌无力的临床证据;(4)皮肤病变出现后2年内肌酶水平均正常[1,2,3,58,82,83,84]。
好发于老年人,男性多于女性。均为隐袭缓慢发病,病程长达1年以上,甚至达数年至10余年。主要表现为缓慢持续性进展的无痛性肌萎缩和肌无力。大多数患者是无意中发现手肌无力,骨间肌萎缩或蹲起困难而就诊。肌无力的特点为进行性非对称性,以股四头肌、屈指肌、屈腕肌和足背屈肌无力显著,后期可累及球部肌肉而出现球麻痹,甚至累及呼吸肌引起呼吸困难。没有感觉障碍和皮肤受损表现。极少合并自身免疫性疾病,无其他组织器官受累表现;也极少合并恶性肿瘤。本病必须经肌肉活检方能明确诊断[1,2,3,24,32,77,78,79,80]。
也可称为免疫性NM(autoimmune necrotizing myopathy,ANM)或坏死性免疫性肌病(necrotising autoimmune myopathy,NAM)。NM是一个病理诊断,由免疫介导引起者称为IMNM,其包括某些特殊抗体阳性的IIM,如抗SRP抗体阳性NM、抗HMGCR抗体阳性坏死肌病;也可没有抗体阳性的NM;还可有继发原因引起的NM,如副肿瘤性NM和结缔组织病相关性NM。IMNM可见于不同年龄,可呈急性、亚急性或慢性发病,均表现为进行性双下肢无力,继而发展为双上肢无力和球麻痹。可查出恶性肿瘤或结缔组织病等相关疾病,或血液某些特异性抗体阳性。该病必须依赖肌肉病理和相关抗体检测结果来进行诊断[1,2,3,24,32,33,45,46,47,48,52,81,85]。
除了以上5个常见类型的IIM外,还有与MSAs相关的其他IIM,如ASS、抗SRP抗体阳性NM或SRP抗体肌病、抗HMGCR抗体阳性的IMNM和抗MDA5抗体阳性ADM,已在上述相关抗体中谈及,在此不再重复。
IIM的诊断与鉴别诊断程序主要有以下内容。
依据患者呈急性或亚急性发病,伴进行性加重的肌无力,通过体检明确四肢近端为主的肌无力,如再伴有咽喉肌无力者,可能为肌肉疾病。如询问出和查出有特殊的皮疹,则对皮肌炎的诊断非常重要。如发现有肺部和心脏病变,则支持IIM的可能。
凡是疑似肌肉病者,应检测血清肌酶。尤其是若肌酸激酶明显增高,则为肌病的可能性大,应进行下一步检查。但肌酸激酶正常者也不能除外某些肌病,如sIBM,其肌酸激酶水平可以为正常。肌酸激酶还可以作为肌炎治疗的观察指标,尤其是治疗后,肌酸激酶的变化远较肌无力改善快。但在治疗减量或停止用药后,如肌酸激酶明显又重新升高,则提示肌炎复发。
肌电图检查是判断是否为肌病或肌肉受损的关键技术。如果可靠的肌电图结果提示有肌源性受损,则可基本判断患者存在肌病,并考虑给予进一步检查或治疗。但值得注意的是,肌电图技术是辅助检查中最为复杂、操作要求很高的技术,许多医院的肌电图技术常不过关,给出的报告并不准确,易导致医生的误判。
凡是疑似IIM的患者,都应进行血清MSAs和MAAs检测。MSAs阳性者可协助判断IIM的类型。
核磁共振为一种无创性技术,可协助判断四肢肌肉、咽喉肌、脊旁肌、腹部肌肉、肋间肌、膈肌等部位是否有病变。IIM病变在MRI上显示出不同程度的高信号,但肌肉明显萎缩后的脂肪化也可呈高信号。磁共振技术不是IIM患者的必须检查技术,其仅可作为IIM的早期筛查或特殊部位肌肉受累的检查。
只要考虑患者有IIM的可能,都应进行肌活检,以协助诊断本病及进行分类;特别是疑似IMNM或sIBM者,必须通过肌活检方能诊断。肌活检必须进行酶组织化学和必要的免疫组织化学检查,必要时还应送电镜检查,以便协助鉴别IIM的类型及其他非炎性肌病的判断。
对于拟诊为IIM的青少年,尤其对于临床表现不典型或肌炎抗体检查阴性或治疗效果不佳且肌活检又不能完全明确诊断者,应送检血或肌肉标本作基因筛查,以除外基因突变相关的非炎性肌病。
IIM是可以治疗性疾病,不论是哪个亚型,其发病形式均可能为急性、亚急性或慢性发病;但最终均发展为慢性过程。因此,在早期均应该积极尽早、足量给予抗免疫药物治疗,以控制病情发展;病情得到有效控制后,应长期应用有效维持量,以达到长期控制病情稳定,最终达到治愈的目标,减少后遗症。对于发病年龄大或治疗效果不理想的患者,应注意除外恶性肿瘤相关的IIM,找到发病原因,针对病因进行治疗。
糖皮质激素为各类IIM的首选药物,根据病情急缓和严重程度的不同,选用直接口服醋酸泼尼松或静脉注射甲泼尼龙琥珀酸钠。(1)醋酸泼尼松片(泼尼松片):病情不严重者可直接口服泼尼松片1.0~1.5 mg·kg-1·d-1,晨起顿服,持续4~5周后开始递减,每1~2周减5 mg,直至减到20~10 mg作为维持量。(2)甲泼尼龙琥珀酸钠:病情严重者给予1 000 mg或500 mg,静脉滴注,1次/d;3 d后减半量,而后再依次隔3 d减半量,直至相当于泼尼松片的初始口服量时改为口服,并按上方法逐渐减量。
当口服药减至维持量后,可以长期服用,一般均在2年以上;短者2~3年,长者达10年以上方可治愈。在治疗和减药的过程中,主要观察临床表现和血肌酸激酶变化,以判断病情。在减药过程中,如果患者出现病情加重或血肌酸激酶明显升高,则应再重新按初始办法用药。
在治疗过程中,注意应用辅助药物防治不良反应,如补钾、补钙和胃黏膜保护剂,并监测血糖、血脂和血压。在治疗过程中,如果病情没有明显好转,应该继续排查恶性肿瘤的可能或采用以下治疗办法。
部分患者对糖皮质激素不敏感或耐受性差或病情较重,可给予IVIg,每次0.4 mg·kg-1·d-1,静脉滴注,连续3~5次,每月一个疗程,可连续3~5个月。
激素治疗效果不佳或病情迅速加重者,可加用免疫抑制剂。可选择的免疫抑制剂有硫唑嘌呤、环磷酰胺、环孢菌素A、氨甲喋呤、硫酸羟氯喹、吗替麦考酚酯等,因IIM的类型及病情不同,可选择不同的免疫抑制剂。在用药过程中注意相关的不良反应及处理。
病情严重且激素治疗无效者,甚至加上静脉用免疫抑制剂后仍无明显效果时,有条件可行血浆置换治疗,也许能有很好的效果。但对于刚用过IVIg者,不考虑再行血浆置换。采用血浆置换者,应告知患者可能并发感染的风险。
这类单抗类药物在国内外均用于治疗结缔组织病或恶性肿瘤,但是越来越多的临床试验提示对IIM治疗有一定效果,不过均为小样本的经验性治疗,效果报道不一。且此类药物价格较贵,建议严重的IIM患者酌情选用。目前能用于治疗IIM的单抗类生物制品有:利妥昔单抗(rituximab)、英夫利昔单抗(infliximab)、阿巴特普单抗(abatacept或阿巴西普单抗)、依那西普单抗(etanercept或enbrel恩利)、依库珠单抗(eculizumab,或艾库组单抗soliris)、阿仑单抗(alemtuzumab/lemtrada/campath)、托珠单抗(tocilizumab,也称塔西单抗)、双马单抗(bimagrumab)、鲁索利替尼单抗(ruxolitinib)、戈利木单抗(simponi或golimumab)。
除了sIBM和晚期癌性相关肌炎外,其他类型的IIM患者只要早期及时地准确诊断,给予早期足量和长期有效维持量的药物治疗,应该有比较好的效果,结局良好。早期恶性肿瘤相关的IIM患者,如果给予有效的恶性肿瘤治疗,预后也很好。迄今为止,sIBM仍缺乏有效的治疗办法。
作者声明不存在利益冲突
None declared
1.1975年Bohan和Peter提出特发性炎性肌病分类是?
A.①原发性多发性肌炎(PM),②原发性皮肌炎,③合并血管炎的儿童PM或皮肌炎,④合并肿瘤的PM或皮肌炎,⑤合并其他结缔组织病的PM或皮肌炎
B.①原发性PM,②原发性皮肌炎,③继发性PM或皮肌炎,④合并肿瘤的PM或皮肌炎,⑤合并其他结缔组织病的PM或皮肌炎
C.①原发性PM或皮肌炎,②继发性PM或皮肌炎,③合并血管炎的儿童PM或皮肌炎,④合并肿瘤的PM或皮肌炎,⑤合并其他结缔组织病的PM或皮肌炎
D.①原发性PM,②原发性皮肌炎,③儿童皮肌炎,④合并肿瘤的PM或皮肌炎,⑤继发性PM或皮肌炎
E.以上都不对
2.2004年欧洲神经肌肉病中心(ENMC)对特发性炎性肌病又提出了新的分类是?
A.原发性PM或皮肌炎、继发性PM或皮肌炎、散发性包涵体肌炎(sIBM)、非特异性肌炎和免疫性坏死性肌病
B.原发性PM或皮肌炎、继发性PM或皮肌炎、sIBM和非特异性肌炎
C.原发性PM或皮肌炎、继发性PM或皮肌炎、sIBM和免疫性坏死性肌病
D.原发性PM或皮肌炎、继发性PM或皮肌炎、sIBM和坏死性肌病
E.PM、皮肌炎、sIBM、非特异性肌炎和免疫介导的坏死性肌病
3.肌炎特异性抗体主要包括?
A.抗Jo-1抗体、抗U1snRNP、抗SRP抗体、抗MDA5抗体、抗HMGCR抗体、抗RNP抗体和抗PM-Scl抗体
B.抗Jo-1抗体、抗U1snRNP、抗SRP抗体、抗MDA5抗体、抗HMGCR抗体、抗RNP抗体和抗ANA抗体
C.抗ARS抗体、抗U1snRNP、抗SRP抗体、抗MDA5抗体、抗HMGCR抗体、抗RNP抗体和抗PM-Scl抗体
D.抗ARS抗体、抗SRP抗体、抗MDA5抗体、抗HMGCR抗体、抗Mi-2抗体和抗NXP-2抗体
E.抗ARS抗体、抗SRP抗体、抗MDA5抗体、抗HMGCR抗体、抗Mi-2抗体和抗U1snRNP抗体
4.以下哪项是散发性包涵体肌炎的诊断关键点?
A.磷酸肌酸肌酶
B.肌肉酶组织化学
C.临床表现
D.免疫组织化学
E.肌肉MRI
5.以下哪项是错误的?
A.皮肌炎患者肌肉病理特征是坏变萎缩肌纤维呈束周分布
B.坏死性肌病的诊断取决于肌肉活检结果
C.多发性肌炎都好发于各个年龄段
D.散发性包涵体肌炎的诊断取决于肌肉活检结果
E.特发性炎性肌病的治疗首选药物是糖皮质激素

























