
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
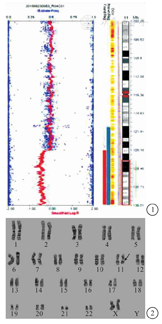
孕妇 30岁,孕1产0,非近亲结婚,否认家族遗传病史,无有害物质接触史。孕11+5周超声示NT=0.10 cm。孕18+2周血清学筛查提示21三体高风险(1/235)。在征得患者知情同意[伦理审查批准号:(2018)临审(2018-379)号]后,于18+6周行羊膜腔穿刺术,采集两份羊水细胞标本,一份行人类全基因组SNP分型芯片(Human CytoSNP-12 DNA Analysis BeadChip,美国Illumina公司)检查,结果显示arr[hg19]11q24.2q25 (125 281 236-134 944 006) ×1,提示胎儿染色体11q24.2q25区存在约9.66 Mb缺失(图1)。另一份常规进行羊水细胞培养,收获,制备染色体,G显带,计数50个分裂相,参照人类细胞遗传学国际命名体制(ISCN 2016)进行染色体核型描述,胎儿染色体核型为46,XN,del(11)(q24.2q25)(图2)。孕妇及其配偶染色体核型均未见异常。孕妇签署知情同意书,并经过医院伦理委员会批准(临审第2018-379号)。
综合aCGH检测及核型分析的结果,胎儿的双亲最终选择在23+1周中止妊娠。孕妇在被注射利凡诺后,于次日自然分娩一死胎,后者身长24 cm。胎盘自然娩出,外观完整,表面粗糙,胎膜缺损1/2。引产儿由于孕周较小,未发现其存在明显的外部畸形。
11号染色体长臂末端缺失综合征又称Jacobsen综合征(Jacobsen sydrome,JBS),是一种罕见的11q远端缺失导致的神经发育迟缓与多发畸形的综合征[1]。约70%的JBS患者为女性,这可能是由于11q末端的基因在某种程度上影响了性别的发育。目前已报道的JBS的缺失范围为5~20 Mb,关键区域位于11q23-11qter,且以末端缺失为主。本例胎儿染色体11q24.2q25区存在约9.66 Mb缺失,缺失片段位于JBS综合征的关键区域,共涉及51个蛋白编码基因,其中40个被OMIM数据库收录基因。56%的JBS患儿具有先天性心血管系统异常[2],主要表现为房间隔缺损、室间隔缺损、动脉导管未闭、左心梗阻性畸形、肺动脉硬化和双右心室流出道等,其中最常见的包括室间隔缺损和左心梗阻性畸形(主动脉畸形、二尖瓣狭窄、主动脉缩窄、左心发育不全综合征)。本例胎儿缺失区域与心脏发育相关的基因包括JAM3(OMIM 606871)、ETS1(OMIM 164720)、BARX2(OMIM 604823)、 FLI1(OMIM 193067)、NTM(OMIM 607938)、KIRREL3(OMIM 607761)、B3GAT1(OMIM 151290)等。51%的JBS患儿存在头颅MRI的异常如双侧脑室扩大、不对称、脑白质减少、脑萎缩、小脑发育不良和胼胝体发育不良等。本病例与神经系统发育相关的基因包括NRGN(OMIM 602350)、KIRREL3、NTM及B3GAT1。除心脏及神经系统发育异常外,还有小头畸形等特殊面容、短颈、骨骼、肾脏发育异常等。但本例胎儿引产时孕周较小,且孕妇及家人拒绝对其进行病理学检测,因此无法判定引产儿是否有上述表型。对于孕周较小但检出染色体重复或缺失的病例,在遗传咨询时应充分告知其可能导致的疾病,并建议其在再次妊娠时进行产前诊断。
所有作者均声明不存在利益冲突

























