
探讨我国GATA2突变相关儿童原发性骨髓增生异常综合征(MDS)的发生情况、临床特点及分子生物学特征。
回顾性分析2007年1月至2018年1月129例儿童原发性MDS患者临床资料,采用二代测序技术检测GATA2及髓系恶性肿瘤相关基因突变情况。分析基因突变谱及其与临床表现型的关系。
在所有129例患者中,11例(8.5%)检出GATA2胚系突变。在50例MDS伴原始细胞增高(MDS-EB)和急性髓系白血病伴MDS相关改变(AML-MRC)患者中,GATA2胚系突变占14.0%。GATA2突变多位于第二个锌指(ZF2)区。多因素分析结果显示,SETBP1体细胞突变(P=0.041,OR=9.003,95%CI 1.098~73.787)和独立的7号染色体单体(P=0.002,OR=24.835,95%CI 3.305~186.620)与GATA2胚系突变显著相关。与GATA2野生型的患者相比,GATA2突变型患者中位发病年龄更大[8(1~16)岁对6岁(1月龄~18岁),P=0.035],更易伴有7号染色体单体(72.7%对5.2%,P<0.001),较之儿童难治性血细胞减少(RCC)更倾向于存在于MDS-EB/AML-MRC亚型中(5.1%对13.7%,P=0.111)。GATA2突变与否不影响儿童原发性MDS患者的3年预期总生存(OS)率[(80.1±4.2)%对(60.6±25.4)%,P=0.437];在44例接受allo-HSCT患者中,GATA2突变与否对移植后3年预期OS率无显著影响[100.0%对(94.0±3.8)%,P=0.562]。
GATA2突变在我国伴有7号染色体单体及年龄较大的儿童原发性MDS患者中占有较高的比例,且GATA2突变患者多进展为MDS-EB和AML-MRC。GATA2突变状态不影响儿童原发性MDS的总体生存。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
骨髓增生异常综合征(MDS)是一组具有异质性的克隆性造血性疾病,儿童罕见。在儿童和青少年中,儿童难治性血细胞减少(refractory cytopenia of childhood,RCC)是最常见的亚型,常表现为全血细胞减少和低增生骨髓象[1,2]。儿童MDS最常见的染色体核型异常是7号染色体单体(-7/7q-),而在成人中常见的单纯5号染色体缺失(5q-)和复杂核型罕见[3]。儿童和成人MDS体细胞突变谱亦显著不同[4,5,6]。MDS可继发于先天性骨髓衰竭(IBMF),如范可尼贫血、先天性角化不良及家族性MDS/急性髓系白血病(AML)综合征。其中家族性MDS/AML综合征是由造血转录因子CEBPA、RUNX1和GATA2等缺陷导致的[7]。GATA2相关的疾病谱还包括免疫缺陷和血液系统以外的综合征,如MonoMAC综合征和Emberger综合征等[8]。GATA2突变患者在婴儿或儿童期即出现造血、免疫和淋巴系统症状,并伴有反复感染,高风险进展为MDS/AML[9,10]。目前我国尚无儿童MDS胚系GATA2突变情况的研究,这给实验室检查选择、预后评估及治疗决策带来一定的困难。本研究探讨GATA2胚系突变在我国儿童原发性MDS中的发生频率,并分析GATA2相关儿童MDS的临床及分子生物学特征。
回顾性分析我院2007年1月至2018年1月确诊的129例儿童原发性MDS患者,MDS诊断符合2016年WHO分型诊断标准儿童MDS诊断标准[11]。包括RCC 78例(61%)、MDS伴原始细胞增高(MDS-EB)35例(27%)和急性髓系白血病伴MDS相关改变(AML-MRC)16例(12%)。排除继发性MDS。本研究经我院伦理委员会审核批准,并获得所有研究对象及(或)其监护人的知情同意。患者根据病情及治疗意愿接受免疫抑制治疗、化疗、allo-HSCT或支持治疗[12]。其中,34%(44/129)的患者接受了allo-HSCT。供者来源:同胞全相合3例,父母/同胞不全相合41例。移植方式:16%(7/44)为外周血+骨髓,82%(36/44)为外周血,2%(1/44)为外周血+骨髓+脐带血。11例GATA2突变患者中7例接受了HSCT。
骨髓及体细胞对照标本取自患者初次诊断时。常规方法留取骨髓单个核细胞,提取DNA。体细胞对照DNA提取自初诊时的口腔上皮细胞。家系验证DNA提取自父母和(或)同胞外周血DNA。
设计包含与IBMF、家族性MDS/AML、MDS相关的基因等在内的564个基因的全部外显子及剪接区的捕获芯片。取1 μg DNA常规方法制备DNA全基因组文库。采用GenCap液相捕获试剂盒(北京迈基诺基因科技股份有限公司产品)对文库目标区域进行捕获及定量,采用Illumina Nextseq 500测序。平均覆盖度98.1%,平均深度1 000×,95%的目的区域覆盖度在20×以上。测序后原始数据利用CCDS、人基因组数据库(HG19)、dbSNP(v138)、1 000 genomes、COSMIC、PolyPhen、SIFT等数据库进行生物信息学分析,确定致病性基因突变位点[13],并采用Sanger测序验证。新发GATA2变异依据以下标准判断为可能致病性和疾病相关[14]:①其他患有MDS的家庭成员具有同样的突变;②具有其他GATA2缺陷相关的表型(如淋巴水肿);③既往在GATA2缺陷中报道过的突变;④在Exome Aggregation Consortium Browser、dbSNP和Exome Variant Server数据库中未被报道的新发突变;⑤在物种间高度保守及预测致病性高的位点。骨髓细胞经过24 h培养,收集细胞常规制片,G或R显带,根据《人类细胞遗传学国际命名体制(ISCN2013)》描述核型异常。单纯7号染色体单体指经染色体核型分析及FISH检测,除7号染色体单体外不伴有其他异常核型;涉及7号染色体单体指出7号染色体单体外还伴有其他异常核型。
所有病例随访至2018年10月。随访资料来源于住院病历、门诊病历。对随访期间死亡的病例,依病历记录或与患者家属电话联系确认。预期总生存(OS)期为确诊至死亡的时间或随访终点。共9例(7%)患者失访。中位随访时间为29(2~131)个月。
采用SPSS 22.0软件进行统计学分析。非连续变量资料采用卡方检验或者Fisher精确概率法,连续变量资料采用Mann-Whitney U检验,相关性分析采用Spearman等级相关系数,单因素生存分析采用Kaplan-Meier法,将单因素分析P<0.01的指标纳入Cox模型进行多因素分析。P<0.05为差异有统计学意义。
8.5%(11/129)的儿童原发性MDS患者具有GATA2突变。较之RCC,GATA2在MDS-EB/AML-MRC中的比例更高(5.1%对13.7%,P=0.111)。GATA2突变型患者首次诊断年龄晚于GATA2野生型患者[8(1~16)岁对6岁(1月龄~18岁),P=0.035]。GATA2突变患者更易合并7号染色体单体(72.7%对5.2%,P<0.001)。因为RCC诊断标准宽泛,我们对其中同时符合成人MDS伴多系血细胞发育异常(MDS-MLD)诊断标准[11]的患者进行分析,发现所有的GATA2突变患者均属于此组。所有的GATA2突变患者均为自发突变,无血液系统恶性肿瘤家族史。与GATA2野生型患者相比,GATA2突变患者血小板计数更高[100(17~299)×109/L对36.5(2~528)×109/L,z=-2.058,P=0.040]。其他实验室检查指标(外周血血红蛋白浓度、白细胞计数、中性粒细胞计数、单核细胞计数及骨髓髓系原始细胞比例)无显著性差异(表1)。
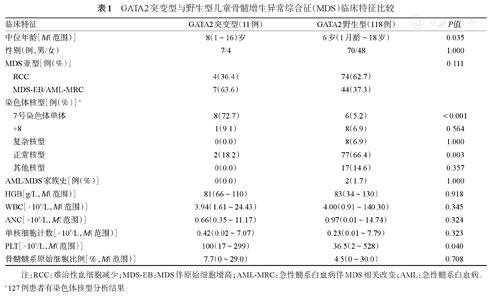
GATA2突变型与野生型儿童骨髓增生异常综合征(MDS)临床特征比较
GATA2突变型与野生型儿童骨髓增生异常综合征(MDS)临床特征比较
| 临床特征 | GATA2突变型(11例) | GATA2野生型(118例) | P值 | |
|---|---|---|---|---|
| 中位年龄[M(范围)] | 8(1~16)岁 | 6岁(1月龄~18岁) | 0.035 | |
| 性别(例,男/女) | 7/4 | 70/48 | 1.000 | |
| MDS亚型[例(%)] | 0.111 | |||
| RCC | 4(36.4) | 74(62.7) | ||
| MDS-EB/AML-MRC | 7(63.6) | 44(37.3) | ||
| 染色体核型[例(%)]a | ||||
| 7号染色体单体 | 8(72.7) | 6(5.2) | <0.001 | |
| +8 | 1(9.1) | 8(6.9) | 0.564 | |
| 复杂核型 | 0(0.0) | 8(6.9) | 1.000 | |
| 正常核型 | 2(18.2) | 77(66.4) | 0.003 | |
| 其他核型 | 0(0.0) | 17(14.6) | 0.357 | |
| AML/MDS家族史[例(%)] | 0(0.0) | 2(1.7) | 1.000 | |
| HGB[g/L,M(范围)] | 81(66~110) | 83(34~130) | 0.918 | |
| WBC[×109/L,M(范围)] | 3.94(1.61~24.43) | 4.00(0.91~140.30) | 0.345 | |
| ANC[×109/L,M(范围)] | 0.66(0.35~11.17) | 0.97(0.01~14.74) | 0.324 | |
| 单核细胞计数[×109/L,M(范围)] | 0.42(0.02~7.07) | 0.23(0.01~7.79) | 0.323 | |
| PLT[×109/L,M(范围)] | 100(17~299) | 36.5(2~528) | 0.040 | |
| 骨髓髓系原始细胞比例[%,M(范围)] | 7.7(0~29.0) | 4.5(0~30.0) | 0.708 | |
注:RCC:难治性血细胞减少;MDS-EB:MDS伴原始细胞增高;AML-MRC:急性髓系白血病伴MDS相关改变;AML:急性髓系白血病。a 127例患者有染色体核型分析结果
11例伴有GATA2突变的MDS患者中,染色体核型异常包括7号染色体单体8例,其中非平衡易位der(1;7)(q10;p10)(导致7号染色体长臂缺失)2例;8号染色体三体1例;未检测到-5/5q-及复杂核型。在127例具有可分析染色体核型的患者中,7号染色体单体14例(11.0%),而其中8例具有GATA2突变,且诊断时的中位年龄倾向于大于6例GATA2野生型患者[11(3~16)岁对5.5(1~10)岁]。
共在11例儿童MDS患者中检测到12个致病性或可疑致病性胚系突变(其中10个为新发突变),及1个意义未明突变。包括7个移码/无义突变导致的截短突变,3个错义突变,1个非移码缺失突变,1个剪接位点突变。等位基因突变频率在0.28~0.62之间。11个突变位于编码区,其中6个突变位于第二个锌指(ZF2)区(氨基酸347~398),2个位于C末端,1个位于N末端。位于非编码区的突变c.1143+2T>G位于第5内含子,邻近剪接位点。其中1例患者(例10)具有3个突变,包括位于第5内含子的c.1143+2T>G,位于ZF2区的c.1086_1087ins63(p.N36delins22)及位于ZF1区的意义未明的突变c.961C>T(p.L321F)(表2)。GATA2突变类型与血液学表型无相关性。所有患者的父母和(或)正常同胞采用Sanger测序进行了相应位点的检测,未检测到健康携带者。具有GATA2突变的患者中,最常见的体细胞突变为SETBP1(4个)、ASXL1(4个)及RUNX1(3个)突变,5例患者未检出合并体细胞突变。另有1例患者合并SAMD9突变。单因素分析提示,体细胞SETBP1(P=0.001,χ2=10.438)、ASXL1(P<0.001,χ2=13.766)、RUNX1(P=0.002,χ2=9.179)突变,单纯7号染色体单体(P<0.001,χ2=46.748),涉及7号染色体单体(P<0.001,χ2=26.229)与GATA2突变显著相关。进一步多因素分析提示,SETBP1突变(P=0.041,OR=9.003,95%CI 1.098~73.787)和单纯7号染色体单体(P=0.002,OR=24.835,95%CI 3.305~186.620)与GATA2突变显著相关。
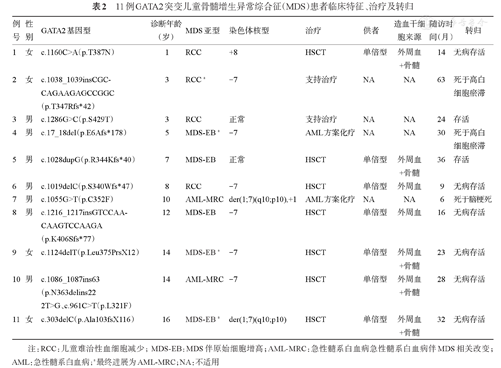
11例GATA2突变儿童骨髓增生异常综合征(MDS)患者临床特征、治疗及转归
11例GATA2突变儿童骨髓增生异常综合征(MDS)患者临床特征、治疗及转归
| 例号 | 性别 | GATA2基因型 | 诊断年龄(岁) | MDS亚型 | 染色体核型 | 治疗 | 供者 | 造血干细胞来源 | 随访时间(月) | 转归 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 女 | c.1160C>A(p.T387N) | 1 | RCC | +8 | HSCT | 单倍型 | 外周血+骨髓 | 14 | 无病存活 |
| 2 | 女 | c.1038_1039insCGC-CAGAAGAGCCGGC(p.T347Rfs*42) | 3 | RCC a | -7 | 支持治疗 | NA | NA | 63 | 死于高白细胞瘀滞 |
| 3 | 男 | c.1286G>C(p.S429T) | 3 | RCC | 正常 | 支持治疗 | NA | NA | 24 | 存活 |
| 4 | 男 | c.17_18del(p.E6Afs*178) | 5 | MDS-EB a | -7 | AML方案化疗 | NA | NA | 30 | 死于高白细胞瘀滞 |
| 5 | 男 | c.1028dupG(p.R344Kfs*40) | 7 | MDS-EB | 正常 | HSCT | 单倍型 | 外周血+骨髓 | 36 | 存活 |
| 6 | 男 | c.1019delC(p.S340Wfs*47) | 8 | RCC | -7 | HSCT | 单倍型 | 外周血 | 9 | 无病存活 |
| 7 | 男 | c.1055G>T(p.C352F) | 10 | AML-MRC | der(1;7)(q10;p10),+1 | AML方案化疗 | NA | NA | 6 | 死于脑梗死 |
| 8 | 男 | c.1216_1217insGTCCAA-CAAGTCCAAGA(p.K406Sfs*77) | 12 | MDS-EB | -7 | HSCT | 单倍型 | 外周血 | 16 | 无病存活 |
| 9 | 女 | c.1124delT(p.Leu375PrsX12) | 14 | MDS-EB a | -7 | HSCT | 单倍型 | 外周血+骨髓 | 23 | 无病存活 |
| 10 | 男 | c.1086_1087ins63(p.N363delins222T>G、c.961C>T(p.L321F) | 14 | AML-MRC | -7 | HSCT | 单倍型 | 外周血+骨髓 | 28 | 无病存活 |
| 11 | 女 | c.303delC(p.Ala103fsX116) | 16 | MDS-EB a | der(1;7)(q10;p10) | HSCT | 单倍型 | 外周血+骨髓 | 32 | 无病存活 |
注:RCC:儿童难治性血细胞减少;MDS-EB:MDS伴原始细胞增高;AML-MRC:急性髓系白血病急性髓系白血病伴MDS相关改变;AML:急性髓系白血病;a最终进展为AML-MRC;NA:不适用
除1例(例5)具有p.R344Kfs*40突变的患者同时合并先天性淋巴水肿外,1例(例6)具有p.S340Wfs*47突变的患者合并先天性上睑下垂外,其他患者均无非血液系统表型。
全部患者3年预期OS率为(79.1±4.3)%。RCC患儿的3年OS率显著高于MDS-EB/AML-MRC患者[(91.1±4.0)%对(59.9±8.7)%,P<0.001]。单因素分析示,GATA2突变型与GATA2野生型患者3年OS率差异无统计学意义[(80.1±4.2)%对(60.6±25.4)%,P=0.437](图1A)。44例行allo-HSCT患者的3年OS率显著高于未行移植的患者[(95.2±3.3)%对(77.3±5.0)%,P=0.002](图1B)。在接受移植治疗患者中,GATA2基因突变状态对移植后3年OS率无显著影响[100.0%对(94.0±3.8)%,P=0.562]。
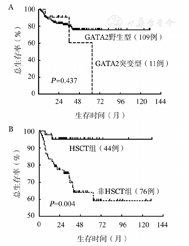
A:GATA2突变型与GATA2野生型;B:造血干细胞移植组(HSCT)组与非HSCT组
近年来遗传易感性在原发性MDS中的作用受到关注。本研究结果示在我国儿童原发性MDS中,GATA2突变占到8.5%。而在MDS-EB和AML-MRC的患者中,占到13.7%。这与既往文献[14]中报道的比例相似。而成人MDS患者中,GATA2突变的发生率仅为0.5%,GATA2突变相关疾病的中位发病年龄为29岁[9],而针对儿童及青少年的研究中,GATA2相关MDS的中位发病年龄为12岁[14],结合本研究结果及文献报道,推测GATA2相关疾病主要存在于青少年及年轻成人MDS患者中,GATA2不是老年患者的主要致病基因。
既往报道过的与GATA2突变有关的综合征包括Emberger综合征(原发性淋巴水肿、皮肤疣、耳聋)、MonoMAC综合征(严重的单核细胞、NK细胞、树突状细胞和B淋巴细胞减少,鸟结核分枝杆菌复合物感染)[8],非血液系统表型包括性功能减退、外眦赘皮、手指变细、颈蹼、行为障碍/多动症、甲状腺功能减退、泌尿生殖系统畸形、单侧/双侧上睑下垂或胎儿水肿[15]。值得注意的是,本研究中GATA2相关的MDS均为散发,且大部分患者无GATA2缺陷相关的综合征或非血液系统的表型。而接近半数的GATA2相关非MDS疾病伴有综合征相关症状[14,16]。在儿童MDS中高比例的新发GATA2突变及极少的无症状携带者表明该基因突变较高的外显率和表现度。本研究GATA突变发生率高,且编码区突变具有完全的外显率,但因为病例数量有限,可能存在偏倚。既往研究曾报道在超过200例患者中检测到8例无症状携带者[9]。此外,非编码区突变的无症状携带者,外显率减低既往也有报道[8,14]。
GATA2基因编码重要的造血转录因子,其通过两个ZF与上千个基因的GATA结合结构域作用[17]。GATA2以剂量依赖的方式,通过与其他转录因子协同作用,调控早期造血[18]。GATA2胚系突变涉及截短突变多导致ZF2缺失[8,15]。此外,目前认为ZF2内的错义突变和位于GATA2-9.5 kb调控区的非编码区变异导致半倍体剂量不足[7,8,15]。GATA2体细胞突变也存在于成人髓系肿瘤中,不同的是,突变可发生于两个ZF区,既可引起功能获得亦可引起功能缺失效应[15,19]。
与既往报道GATA2半倍体剂量不足导致血细胞减少及免疫缺陷相似,我们检测到2种单等位基因突变,包括截短突变和位于ZF2区的错义突变。但未检测到位于第4内含子的增强子区域突变。迄今为止,只有一小部分的GATA2错义突变进行了功能验证。丧失功能性突变包括在家族性MDS/AML中位于ZF2区的Thr354Met及Thr355del13,在Emberger综合征中的Arg361Leu及Cys373Arg17,在原发性AML中的Arg350_Asn351ins(体细胞突变状况不明)[20]。本研究我们未检测到上述突变。本研究及既往报道GATA2截短突变均破环了ZF2区,提示这一区域的重要性。Wlodarski等[14]研究显示非编码区突变占10.5%,且部分非编码区突变的患者具有更长的疾病稳定期,但本研究只检测到1个内含子区的突变。本研究及既往研究均未发现GATA2基因型与临床表型具有显著相关性。
高危险度MDS亚型和7号染色体单体在GATA2相关MDS中比例显著增加。相反,在GATA2相关免疫缺陷患者中,8号染色体三体(24%)多于7号染色体单体(16%)[8]。既往报道7号染色体单体在整个年龄组GATA2突变中占29%[9],在儿童及青少年组中占70%[17],这与本儿童队列中结果基本一致(72.7%)。而且,本研究中,2例患者为der(1;7)(q10;p10)非平衡易位,导致1q三体和7q缺失,这一变异在儿童MDS中较少见[4,6]。既往研究[14]及本研究均证实GATA2胚系突变在伴有7号染色体单体的青少年MDS患者中比例较高,因此对这部分患者在诊断时需注意除外GATA2突变。
GATA2突变相关疾病易向髓系肿瘤转化的驱动突变尚不清楚。既往研究报道示30%GATA2突变患者可检测到同时合并ASXL1基因突变,且附加的获得性突变与疾病进展有关[21,22]。因此,获得性ASXL1突变被认为与GATA2突变向髓系肿瘤恶性转化有关[9,21,23]。但这一观点目前存在争议,首先,在更大队列的研究中,并未观察到GATA2突变患者具有更高的ASXL1突变比例,其次,之前研究得出的两者之间的联系可能是由于7号染色体单体造成的偏倚[6, 23]。其他GATA2相关MDS常见的突变有SETBP1功能获得性突变。本研究中,多因素及单因素分析均示SETBP1突变与GATA2显著相关,提示这一体细胞突变在GATA2突变的致病机制中具有重要作用。
在考虑了MDS亚型和细胞遗传学因素后,本研究中儿童原发性MDS的OS不受GATA2突变状态的影响,提示MDS亚型是影响预后的主要因素。尽管GATA2突变MDS患者更容易在初次诊断时即为MDS-EB和AML-MRC,但OS与GATA2野生型患者无统计学差异。较大系列(534例MDS患者,其中GATA2相关MDS 57例)的研究[14]亦得出与本研究相似结论:当单因素分析GATA2突变状态时,GATA2突变患者预后较差(5年预期OS率:73%对84%,P<0.05),但当纳入MDS亚型、7号染色体单体共同分析时,GATA2突变状态与OS无显著相关性。因此考虑GATA2缺陷虽具有很强的MDS倾向性,但可能不影响总体生存。这一结论需在更大规模的样本中验证。HSCT是目前唯一可以治愈GATA2相关MDS的治疗。既往NIH研究报道,GATA2突变(包括了家族性MDS/AML,肺泡蛋白沉积或反复感染)患者移植后4年OS率为54%[8]。一项针对儿童GATA2相关MDS的研究报道,HSCT后5年OS率为66%[14]。本研究中GATA2相关MDS患者的预后与上述报道相似。儿童GATA2相关原发性MDS移植后总体生存较好,可能与移植年龄较小及无GATA2相关非血液系统并发症有关。
总之,本研究表明GATA2在我国儿童原发性MDS中发生率约9%,在青少年伴有7号染色体单体的患者中占到72.7%。GATA2突变状态不影响MDS患儿的预后。与所有MDS患儿一样,应根据已知的危险因素,来决定GATA2相关MDS中HSCT的时机和预处理方案。早期诊断GATA2缺陷可以使患者的疾病监测更具有针对性,并减少了非治愈性治疗的应用,尤其是避免免疫抑制治疗。我们建议对所有儿童和年轻成人MDS患者都应进行GATA2基因突变分析,无论其是否具有家族史或者GATA2缺陷相关表型。

























