
报道1例先天性肝纤维化伴先天性肝内胆管扩张症,在临床上罕见,具有重要的诊断参考意义。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性肝纤维化(congenital hepatic fibriosis,CHF)伴先天性肝内胆管扩张症(Caroli病)是一组临床少见的常染色体隐性遗传病,以儿童、青少年多发,属于罕见病例[1]。既往报道病例多以不明原因肝硬化为首发表现就诊,本例患者以肝脏占位就诊,最终经病理及基因学检测证实,现报告如下。
男性患者,15岁,因"间断上腹部不适,发现腹部包块1年余"于2016年4月11日入院。患者1年余前无明显诱因间断出现上腹部不适,每月3~4次,每次持续几小时或几天不等,无明显规律,与进食无关,于剑突下可触及一质软包块,无压痛,未诊治,4天前为明确诊断来我院门诊,行腹部强化磁共振检查显示:肝左外叶局限性增大,符合增生性病变MR表现,脾大,双肾多发囊肿,见图1。今为求进一步诊治来我院,门诊以"肝脏占位"收入我院肝胆外科,患者自发病以来,饮食、睡眠、大小便无特殊,体质量无明显变化。既往史、个人史均无特殊。家族史:其母亲腹部CT:肝脏多发囊肿,双肾多发囊肿,点状血肿可考虑。体格检查:查体未见明显异常,移动性浊音(-),Murphy征(-),肠鸣音3~4次/min。入院后查血常规尿常规、大便常规正常;肝功能指标:丙氨酸氨基转移酶66 U/L,天冬氨酸氨基转移酶53 U/L,γ-谷氨酰转移酶为56 U/L;碱性磷酸酶384 U/L,白蛋白41.6 g/L,总胆红素7.70 μmol/L;凝血酶原时间13.1 s;甲胎蛋白4.64 ng/ml;降钙素原0.4 ng/ml;C反应蛋白0.02 mg/L;抗平滑肌抗体1∶100(+),抗核抗体、抗线粒体抗体、抗双链DNA等免疫指标均(-);免疫球蛋白未见异常。病毒学检测指标:HBsAg(-);丙型肝炎抗体(-),人类免疫缺陷病毒抗体(-),梅毒抗体(-),EB病毒DNA(-),巨细胞病毒DNA(-);血清铁蛋白、血清铜、铜蓝蛋白、甲状腺功能、心肌酶谱未见异常;尿常规、大便常规均未见异常;心电图、心脏超声未见明显异常;胸片、肺功能正常。
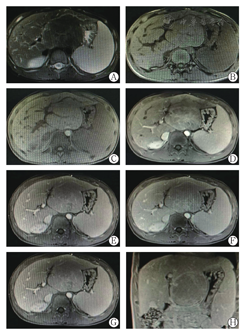
注:肝左外叶见团块状长T1异常信号灶,大小约9.3 cm×6.1 cm×8.6 cm,邻近组织受压移位,注入GD-DTPA后病变与肝实质强化程度相似,其内可见血管走行,胰腺形态及信号未见明显异常,胆囊不大,肝内胆管未见明显扩张,双肾内见多发囊状长T2无强化灶,最大者位于右肾上极,直径2.1 cm,脾大,厚5.1 cm,信号未见明显异常。影像学诊断:(1)肝左外叶局限性增大,符合增生性病变MR表现;(2)脾大;(3)双肾多发囊肿
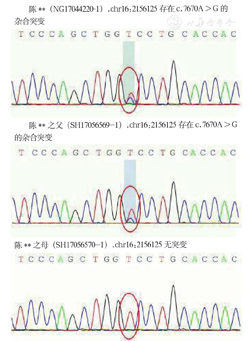
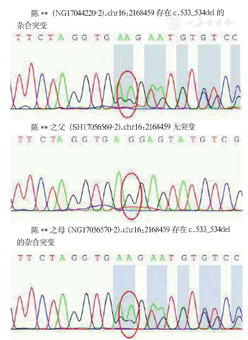
排除手术禁忌,于2016年4月16日在全麻下经腹腔镜行左外叶切除+肝脏肿瘤切除术。术中探查见肝脏表面2个肿物,大者位于左外叶脏面,大小约10 cm×10 cm,小者位于IV段紧邻胆囊处,大小约4 cm×4 cm。手术顺利,术后给予抑酸、保肝、抗感染及对症支持治疗,患者恢复良好出院。病理结果见图2,镜下所见:肝细胞未见明显紊乱,汇管区扩大,宽大致密纤维间隔内见小叶间胆管增生,扩张迂曲排列异常,周围未见炎细胞浸润,界面炎不明显。诊断考虑:CHF伴Caroli病,免疫组织化学检测结果示:细胞角蛋白7(胆管+),细胞角蛋白19(胆管+),干扰素调节因子4(+),铜染色(-),铁染色(-),谷氨酰胺合成酶(灶+),CD34(血管+)。抽取患者及父母的外周血行基因检测,检测方法为:安捷伦外显子芯片捕获+高通量测序,基因检测结果显示:该样本在重点关注肝豆状核变性相关基因筛查中未见明确致病改变;在多囊肾、成人1型相关基因(polycystic kidney disease 1,PKD1)存在两处杂合突变,家系验证结果显示此两处杂合突变分别来自患者父母,见表1;外显子检测结果示:该样本在外显子水平未发现明确和疾病相关的拷贝数变异致病的情况;高通量测序结果见表2;突变位点Sanger图片见图3和图4。测序结果参考美国医学遗传学和基因组学学院(American College of Medical Genetics and Genomics,ACMG)相关指南[2]解读显示:来源于其父亲chr16:2156125的c.7670A > G突变意义未明,来源于其母亲Chr16:2168459的c.533_534del突变为可疑致病基因。

陈**及父母遗传检测结果
陈**及父母遗传检测结果
| 基因 | 突变信息 | 陈** | 陈**之父 | 陈**之母 |
|---|---|---|---|---|
| PKD1 | c.7670A > G chr16:2156125 p.D2557G | 杂合突变 | 杂合突变 | 无突变 |
| PKD1 | c.533_534del Chr16:2168459 p.E178fs | 杂合突变 | 无突变 | 杂合基因 |
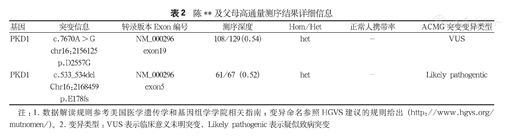
陈**及父母高通量测序结果详细信息
陈**及父母高通量测序结果详细信息
| 基因 | 突变信息 | 转录版本Exon编号 | 测序深度 | Hom/Het | 正常人携带率 | ACMG突变变异类型 |
|---|---|---|---|---|---|---|
| PKD1 | c.7670A > G chr16:2156125 p.D2557G | NM_000296 exon19 | 108/129(0.54) | het | - | VUS |
| PKD1 | c.533_534del Chr16:2168459 p.E178fs | NM_000296 exon5 | 61/67(0.52) | het | - | Likely pathogentic |
注:1.数据解读规则参考美国医学遗传学和基因组学学院相关指南;变异命名参照HGVS建议的规则给出(http://www.hgvs.org/mutnomen/)。2.变异类型:VUS表示临床意义未明突变,Likely pathogenic表示疑似致病突变
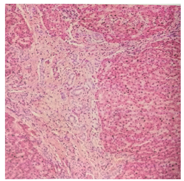
注:肝细胞未见明显紊乱,汇管区扩大,宽大致密纤维间隔内见小叶间胆管增生,扩张迂曲排列异常,周围未见炎细胞浸润,界面炎不明显。诊断考虑:先天性肝纤维化伴Caroli病
截至目前,已随访患者18个月,患者未出现任何不适,术后每隔3个月查1次肝功能、甲胎蛋白、腹部超声,结果均正常。
CHF是常染色体隐性遗传性疾病,临床上以门静脉高压和肝功能正常为特征[3],根据临床表现可分为:门静脉高压型、胆管炎型、混合型和隐匿型,多合并常染色体隐性遗传性多囊肾和或肝内外胆管发育异常,肝组织病理学检查为其确诊的金标准,病理表现通常为纤维间隔内含有形态各异的胆管,可伴有典型的肝内胆管发育畸形或交通性海绵状胆管扩张,即Caroli病[4]。先天性肝内胆管囊性扩张症(又称Caroli病)又称交通性海绵状肝内胆管囊状扩张症或交通性胆管囊肿,是法国学者Caroli于1958年首先报道,是一种较为少见的常染色体隐性遗传性疾病。其临床表现多缺乏特异性,常见症状为:脾肿大、门静脉高压、上消化道出血,早期亦可无明显症状。有观点认为:CHF和Caroli病是同一疾病的不同阶段,CHF伴Caroli病缺乏特征性的临床表现,其隐匿性及复杂性极易导致误诊及漏诊,确诊主要依靠遗传病史及肝活组织学检查。
本病例是以肝脏实性占位为首发表现,最终经病理证实为CHF伴Caroli病,这种情况此前少见报道。该患者基因测序结果示:在多囊肾及成人1型相关基因PKD1存在两处杂合突变,家系验证结果显示此两处杂合突变分别来自患者父母,但患者父母及祖父母均无肝纤维化及胆管扩张表现,只有患者母亲有肝囊肿、肾囊肿存在。测序结果参考美国相关指南[2]解读显示:来源于其父亲chr16:2156125的c.7670A > G突变意义未明,来源于其母亲Chr16:2168459的c.533_534del突变为可疑致病基因。是否该杂合突变就是导致患者致病的原因,目前尚不完全清楚,需要进一步行基础实验加以证实。另外,这两处杂合突变在家系中是以怎样的方式传递,需要进一步将整个家系行基因检测才能明确。CHF伴Caroli病属于罕见病,表现为肝脏实性占位的就更为少见。
到目前为止,该患者术后随访18个月未出现任何不适,对于此类患者手术切除是否能彻底解决问题亦需要长期观察随访。本病例旨在提醒临床及影像科医师对于临床病因不明的肝纤维化及性质不明的占位性病变需要考虑到此病可能,提高大家对临床疑难罕见病例的认识,拓宽诊断思路,提高诊断水平。
所有作者均声明不存在利益冲突

























