
寻找Currarino综合征患儿在已知基因MNX1上的新发突变位点,探索新发突变对原基因与基因产物的结构及功能可能产生的影响,并以其影响为根据阐述Currarino综合征可能的发病机制。
抽取2014~2016年入院治疗的5例Currarino综合征患儿及其核心家属(患儿父母)的外周血液作为样本,应用高通量测序的方法,对5个家族的成员进行外显子测序,通过结果分析,筛选出存在MNX1基因突变的个体,后针对存在MNX1基因突变的个体进行Sanger测序验证,对所得数据结果进行分析,并与已报道的关于Currarino综合征患儿中MNX1突变位点进行对比,寻找新的突变位点。
根据测序与验证试验结果,我们发现了2种发生在外显子上的突变c.863N >T,c.723N >A,其中c.863N >T属于已报道过的突变类型,c.723N >A并未报道过,为新发位点。通过对MNX1基因结构的分析,由于c.723N >A位于基因同源结构域上游,且为终止密码子突变,故该突变的发生对于MNX1基因的功能会产生严重的影响。
目前对于Currarino综合征的基因研究还不能充分解释该病的发病机制,故在已发表的文献中尚无其下行信号传导通路实验及动物模型试验的相关报道,现阶段对于Currarino综合征的研究仍处于基因探索阶段,该综合征的研究仍任重而道远。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Currarino综合征是一种罕见的常染色体显性遗传病,主要症状包括肛门直肠畸形、骶前肿物和骶骨发育畸形。最早由放射学家Currarino于1981年报道[1]。截至目前,诊断方法仍以患儿的症状及体征为主,尚无金标准。但是由于并不是所有的Currarino患儿都具备完整的三联症状,使得Currarino患儿的诊断极为困难。同时目前的基础研究尚不能阐述Currarino综合征基因型和表现型之间的相关性,故发病率难以得到确定。相关学者于1998年第一次确认管家基因MNX1的突变与Currarino综合征的发病有密不可分的关系[2]。如今已有超过80种基因位点的突变或染色体重组得到确认[3]。经过数据归纳分析,在90%的家族性病例中,可以发现MNX1的突变;而在散发病例中,这个概率只有30%[4]。MNX1包含3个外显子,用于编码一个由401个氨基酸组成的蛋白质HB9,这种蛋白质在神经元细胞和胰腺细胞的生长发育中发挥着重要的功能[5]。HB9的氨基酸序列中包含一段由MNX1基因2号和3号外显子编码的同源结构域(第240-300位氨基酸),一段由82个氨基酸组成的高度保守序列(第159-241位氨基酸)和一段重复的丙氨酸序列(第121-134位氨基酸)[6]。本研究纳入了自2014年至2016年首都儿科研究所收治的5例确诊为Currarino三联征的患儿及其父母作为研究对象。收集患儿的下消化道造影、B型超声、MRI或CT的检查结果。同时收集患儿及其家属的血样进行高通量测序。研究发现一种已报的突变类型(c.863N >T,p.Trp288Leu),以及一种新发现的突变类型(c.723N >A,p.Cys241X)。
诊断基于患儿的临床表现、影像学检查和术中组织活检报告。本研究以5个家族作为研究对象。收集每个家族中患儿和父母的外周血液作为样本用于基因检测。
所有患儿的主诉都是肛门的排便功能不良。在询问家族史的过程中,均否认家族中有类似症状的个体存在。入院诊断均以肛门直肠畸形或先天性巨结肠为主要诊断,后经术前影像学检查才确诊为Currarino综合征(其中病例1直至术中发现骶前肿物才被确诊)。
运用经肛门巨结肠根治术对患儿进行手术治疗。5例中有3例为I期手术治疗,2例经历了2次手术(病例2在初次手术后3个月发现吻合口漏,行2次肛门成形术;病例4由于曾在外院行造瘘术,故在我院行关瘘手术)。术中切除瘘管、扩张肠段和骶前肿物来改善肛门直肠的功能。同时将切除的肠段送检,由于病理检查发现了肠壁上的节神经细胞,故排除了先天性巨结肠的可能性。所有患儿术后恢复良好,围手术期没有发生感染、内外出血和肠功能不良的症状。术后1周出院,随访持续进行中(表1)。

5例患儿的临床资料
5例患儿的临床资料
| 编号 | 年龄(月) | 性别 | 骶尾骨畸形 | 骶前肿物 | 肛门直肠畸形 | 其他畸形 | 瘘管 |
|---|---|---|---|---|---|---|---|
| 1 | 8 | 女 | 尾骨缺如 | 成熟型畸胎瘤 | 肛门狭窄 | 双阴道 | 直肠阴道瘘 |
| 2 | 28 | 女 | 镰刀状骶骨,尾骨缺如 | 未成熟型畸胎瘤,脊膜膨出 | 肛门狭窄 | 无 | 无 |
| 3 | 24 | 女 | 尾骨缺如,骶骨部分缺如 | 成熟型畸胎瘤 | 肛门狭窄 | 无 | 直肠会阴瘘 |
| 4 | 21 | 女 | 骶尾骨缺如 | 成熟型畸胎瘤 | 肛门狭窄 | 双阴道 | 直肠会阴瘘 |
| 5 | 2 | 女 | 无异常 | 错构瘤 | 肛门狭窄 | 无 | 直肠会阴瘘 |
由于现阶段的技术原因,送样序列在测序前一般会添加接头,部分测序结果中包含接头序列的信息。此外一次测序常常产生数亿的结果序列,不可避免会出现低质量的测序结果。因此对于序列过滤及质量评估是极为重要的。Fast-QC软件可对测序数据的质量进行整体评估,包括碱基的质量值分布、质量值的位置分布和GC含量等。
应用高通量测序的方法对患儿及其家属的外周血液样本进行基因检测,为了得到测序结果和人类基因组的差异,需将过滤后数据比对到人类基因组(GRCH37.p13)上。然后采用BWA-MEM比对算法(版本:0.7.8-r455)。BWA算法是一个将低发散性序列比对回参考基因组,如人类基因组的先行最佳算法。它包含了三种可选的算法形式:BWA-backtrack、BWA-SW和BWA-MEM。其中BWA-MEM相较于其他两种算法而言,设计更新,更为准确快速,而且在100 bp序列左右的比对区间中的表现比BWA-backtrack更好。
单核苷酸多态性(single nucleotide polymorphism,SNP)以及插入缺失错误分析:基于基因组的比对结果,使用GATK算法以比较每一例样本组织相对于数据库的突变情况。
运用Sanger一代测序验证高通量测序结果的准确性,并根据待测目标片段设计引物,同时进行PCR扩增,若PCR产物测序结果显示待测位点存在套峰,则重新连接T载体测序。将PCR产物连接至pMD -18T载体,化学转化入DH5α菌体内,利用含Amp LB的琼脂平板在37℃的条件下培养菌体。待菌落培养成功后,对单克隆菌落进行PCR验证,并提取质粒进行测序。
将5例患儿及其父母的外周血液进行了外显子测序,结果详见表2。
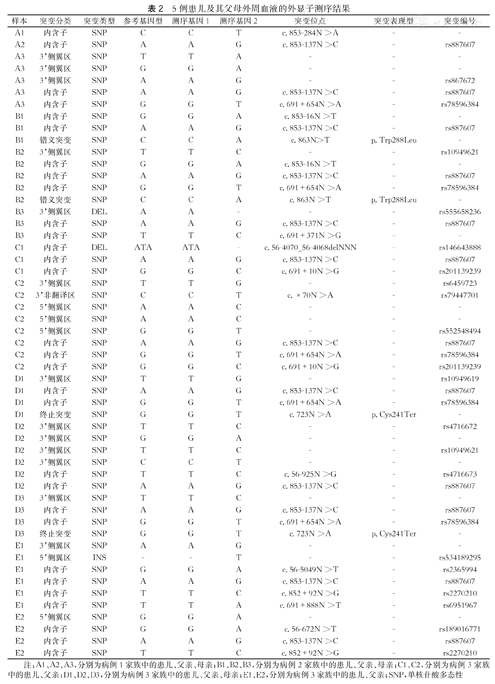
5例患儿及其父母外周血液的外显子测序结果
5例患儿及其父母外周血液的外显子测序结果
| 样本 | 突变分类 | 突变类型 | 参考基因型 | 测序基因1 | 测序基因2 | 突变位点 | 突变表现型 | 突变编号 |
|---|---|---|---|---|---|---|---|---|
| A1 | 内含子 | SNP | C | C | T | c.853-284N >A | - | - |
| A2 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| A3 | 3’侧翼区 | SNP | T | T | A | - | - | - |
| A3 | 3’侧翼区 | SNP | G | G | A | - | - | - |
| A3 | 3’侧翼区 | SNP | A | A | G | - | - | rs867672 |
| A3 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| A3 | 内含子 | SNP | G | G | T | c.691+654N >A | - | rs78596384 |
| B1 | 内含子 | SNP | G | G | A | c.853-16N >T | - | - |
| B1 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| B1 | 错义突变 | SNP | C | C | A | c.863N>T | p.Trp288Leu | - |
| B2 | 3’侧翼区 | SNP | T | T | C | - | - | rs10949621 |
| B2 | 内含子 | SNP | G | G | A | c.853-16N >T | - | - |
| B2 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| B2 | 内含子 | SNP | G | G | T | c.691+654N >A | - | rs78596384 |
| B2 | 错义突变 | SNP | C | C | A | c.863N >T | p.Trp288Leu | - |
| B3 | 3’侧翼区 | DEL | A | A | - | - | - | rs555658236 |
| B3 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| B3 | 内含子 | SNP | T | T | C | c.691+371N >G | - | - |
| C1 | 内含子 | DEL | ATA | ATA | - | c.56-4070_56-4068delNNN | - | rs146643888 |
| C1 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| C1 | 内含子 | SNP | G | G | C | c.691+10N >G | - | rs201139239 |
| C2 | 3’侧翼区 | SNP | T | T | G | - | - | rs6459723 |
| C2 | 3’非翻译区 | SNP | C | C | T | c.*70N >A | - | rs79447701 |
| C2 | 5’侧翼区 | SNP | A | A | C | - | - | - |
| C2 | 5’侧翼区 | SNP | A | A | C | - | - | - |
| C2 | 5’侧翼区 | SNP | G | G | T | - | - | rs552548494 |
| C2 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| C2 | 内含子 | SNP | G | G | T | c.691+654N >A | - | rs78596384 |
| C2 | 内含子 | SNP | G | G | C | c.691+10N >G | - | rs201139239 |
| D1 | 3’侧翼区 | SNP | T | T | G | - | - | rs10949619 |
| D1 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| D1 | 内含子 | SNP | G | G | T | c.691+654N >A | - | rs78596384 |
| D1 | 终止突变 | SNP | G | G | T | c.723N >A | p.Cys241Ter | - |
| D2 | 3’侧翼区 | SNP | T | T | C | - | - | rs4716672 |
| D2 | 3’侧翼区 | SNP | G | G | A | - | - | - |
| D2 | 3’侧翼区 | SNP | T | T | C | - | - | rs10949621 |
| D2 | 3’侧翼区 | SNP | C | C | T | - | - | - |
| D2 | 内含子 | SNP | T | T | C | c.56-925N >G | - | rs4716673 |
| D2 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| D3 | 3’侧翼区 | SNP | T | T | C | - | - | - |
| D3 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| D3 | 内含子 | SNP | G | G | T | c.691+654N >A | - | rs78596384 |
| D3 | 终止突变 | SNP | G | G | T | c.723N >A | p.Cys241Ter | - |
| E1 | 3’侧翼区 | SNP | A | A | G | - | - | - |
| E1 | 5’侧翼区 | INS | - | - | T | - | - | rs534189295 |
| E1 | 内含子 | SNP | G | G | A | c.56-5049N >T | - | rs2365994 |
| E1 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| E1 | 内含子 | SNP | T | T | C | c.852+92N >G | - | rs2270210 |
| E1 | 内含子 | SNP | T | T | A | c.691+888N >T | - | rs6951967 |
| E2 | 5’侧翼区 | SNP | G | G | A | - | - | - |
| E2 | 内含子 | SNP | G | G | A | c.56-672N >T | - | rs189016771 |
| E2 | 内含子 | SNP | A | A | G | c.853-137N >C | - | rs887607 |
| E2 | 内含子 | SNP | T | T | C | c.852+92N >G | - | rs2270210 |
注:A1、A2、A3,分别为病例1家族中的患儿、父亲、母亲;B1、B2、B3,分别为病例2家族中的患儿、父亲、母亲;C1、C2,分别为病例3家族中的患儿、父亲;D1、D2、D3,分别为病例3家族中的患儿、父亲、母亲;E1、E2,分别为病例3家族中的患儿、父亲;SNP,单核苷酸多态性
实验结果处理过程中首先对测序结果进行筛选。Currarino综合征的罕见性决定了其突变基因的突变率在正常人群中不会过高,因此根据Genome 1000及EXAC数据库资料,剔除了突变率>0.1%的突变位点,仅保留了突变率<0.1%或突变率为NONE的突变位点。然后根据外显子测序的质量,排除覆盖倍数不足或测序深度不够的位点。由于现阶段尚不能明确解释内含子突变对外显子的影响,故本次实验中仅对发生于基因编码区可直接编码产物蛋白序列的突变位点进行了分析。最终,确认了2种分别发生于病例2和病例4两个家族的外显子突变。在病例2的检测结果中发现了一种错义突变(c.863N>T)可以引起蛋白产物第288位氨基酸由色氨酸变为亮氨酸,这种突变同样存在于该患儿的父亲检验样本中。而在病例4及其母亲的检测结果中,发现了另一种截止突变(c.723N>A)可以导致蛋白产物第241位氨基酸由半胱氨酸变为终止密码子并丢失其后的全部氨基酸序列。将以上两个家族的DNA样本进行了Sanger一代测序验证,验证了实验结果。
本研究所发现的两种突变类型均发生于同源结构域中,其中c.863N>T、p.Trp288Leu由Hagan等[7]于2000年报道。在病例4发现的c.723N>A、p.Cys241X定位于该同源结构域中的第一位氨基酸,作为截止突变,它的改变不仅意味着该位氨基酸的改变,同时还代表着该基因产物丢失了其后全部同源结构域及之后的序列,相对于Hagan等[7]报道的c.853N>T、p.Trp288Leu(同源结构域中的单一氨基酸突变),我们认为这种突变无疑会带来更为严重的后果。
此外,本研究中存在一些无法解读的问题:①病例2和病例3家族的基因突变均为遗传性,但患儿却均为家族中唯一患病个体,相关文献中也报道过家族性病例的成员中存在着虽伴有突变却不发病的个体[8];②MNX1基因的主要功能在于调控神经元细胞和胰岛细胞发育,与Currarino三联征的表现型之间的关系仍需探讨;③仍存在大量类似于Kim等[9]报道的并无MNX1基因突变信息的病例,如何解释这类人群的发病机制也成为此种疾病研究的重点与难点;④Currarino综合征患儿的症状多变,Cuturilo等[10]发现除三联征的表现多变外,还常伴有智力低下、发育迟缓和面部畸形等症状。
可从以下几方面解决以上问题:①Currarino综合征作为多基因遗传病即其可能受多种基因共同的影响。Holm等[11]通过全外显子测序的方法发现了Currarino综合征新的突变基因ETV3L、ARID5A和NCAPD3。Dworschak等[12]于2017年在一例经一代测序没有发现MNX1基因突变的Currarino综合征患儿的基因检测中发现了新的突变位点dup 3q26.32-q27.2。Lee等[13]也提出Currarino综合征可能受多基因的共同影响。②MNX1基因的突变可能具有组织特异性,即在不同的组织中,突变产生的结果可能不同。③单倍计量不足有可能是导致家族内较少个体发病的原因。在Costamzo等[14]的多中心研究中,研究人员根据Currarino综合征患儿的三联征的症状表现程度分为轻、中、重度共3组,并对MNX1基因进行测序检测,在三种症状全部表达的重度组MNX1基因突变的概率最大,而只表达一种症状的轻度组MNX1基因突变概率最低。杂合突变在现阶段的基因研究中被认为是生物多样性的重要原因之一,故由于杂合突变引起的单倍剂量不足也有可能是家族性病例发病不规律的原因。④多种修饰(表观遗传学)是近年来基因研究的重点,通过对DNA双链的直接修饰改变其表达活性已是最常见的基因表达调控方式。
现在Currarino综合征的治疗方式仍以手术为主[15,16,17]。但相关研究仍存在很多障碍,解决这些障碍最好的方法就是增加病例的数量,但由于其本身是一种罕见的遗传性疾病,发病率低,病例稀少已成为研究该疾病最大的阻碍,而病例数量的积累仍需更长的时间过程。所以我们将继续收集、积累临床病例并处理数据,探索Currarino综合征的发病机制,为临床治疗提供更多的可能性。
所有作者均声明不存在利益冲突

























