
1例主诉为"皮肤发黄6个月余加重3 d"的患儿,综合其临床症状、实验室检查及肝脏病理结果,并完善基因测序示SPTB基因移码突变:c.1791delG,为新发突变。诊断为遗传性球形红细胞增多症。以胆汁淤积起病的遗传性球形红细胞增多症少见。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿男,5岁,因"皮肤发黄6个月余加重3 d"于2019年2月就诊于保定市儿童医院(北京儿童医院保定医院)。入院前6个月,家长发现患儿皮肤和巩膜黄染,无皮肤瘙痒及其他不适,1个月前患儿间断诉腹部及腰背部疼痛,休息后可缓解,3 d前患儿皮肤黄染加重,尿色黄,粪便正常。患儿1月龄时曾诊断"溶血性贫血、黄疸"并输血治疗。患儿为其母第1胎第1产,足月自然出生,智力发育同正常同龄儿。父母非近亲结婚。无家族遗传病史。
体格检查:体温37.1 ℃,脉搏102次/min,呼吸24次/min,血压96/60 mmHg(1 mmHg=0.133 kPa),体重21 kg。双侧巩膜黄染。K-F环阴性。皮肤黄染,无肝掌及蜘蛛痣。腹平,肝脏肋下3 cm,剑突下3 cm可触及,质地中等,脾脏肋下2 cm触及,质地中等,无触痛。
实验室检查(括号内为参考值):肝功能示天冬氨酸转氨酶33.5~64.4 U/L(5~40 U/L),丙氨酸转氨酶12.2~167.1 U/L(5~40 U/L),γ-谷氨酰胺转移酶22.1~117.7 U/L(5~50 U/L),总胆红素229.4~494.6 μmol/L(2~19 μmol/L),直接胆红素154.9~350.5 μmol/L(2~19 μmol/L),间接胆红素74.6~250.6 μmol/L(1.7~13.0 μmol/L)。血常规示白细胞计数(4.8~8.1)×109/L[(4~10)×109/L],红细胞计数(3.4~3.9)×1012/L[(3.5~5.5)×1012/L],血小板计数(277~574)×109/L[(100~300)×109/L],血红蛋白102~110 g/L(110~160 g/L)。尿常规示尿胆原+,尿胆红素++。网织红细胞0.060(0.005~0.015)。红细胞渗透脆性试验阳性。血涂片球形红细胞阴性。嗜肝病毒检测均阴性,肝炎病毒均阴性。血铜、尿铜以及血铜蓝蛋白测定正常。抗核抗体谱均阴性。抗中性粒细胞胞质抗体阴性。直接抗人球蛋白试验阴性。Coombs试验阴性。自身免疫性肝炎抗体阴性。毒检阴性。骨髓片示球形红细胞0.05。腹部彩超示肝右肋下3.5 cm,剑下3.9 cm,脾肋下1.5 cm,实质回声稍增强。胆囊内强回声光点漂浮(首先考虑胆汁淤积)。腹部CT示肝内胆管扩张。腹部磁共振胰胆管造影示肝右叶前段格林森鞘稍增厚,胆汁淤积,脾脏增大。肝脏穿刺病理检查示肝内胆汁淤积性肝病改变(图1)。
基因检查:北京迈基诺医学检验所基因检测结果显示SPTB基因移码突变:c.1791delG(图2),导致氨基酸改变p.Y600Tfs*22,患儿父母均未检测到相关突变,考虑为自发突变,且本突变为新发突变。经过蛋白功能预测软件预测为未知。且未见UGT1A1、ABCC2、SLCO1B3、SLCO1B1等胆红素排泄相关基因上有功能性变异。
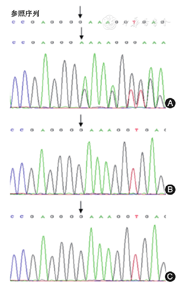
箭头示突变位点
诊疗经过:根据患儿存在溶血性贫血、黄疸病史,入院后查体示肝脾肿大、辅助检查提示贫血、网织红细胞升高、红细胞渗透脆性试验阳性、Coombs试验阴性,结合基因检测结果,诊断为遗传性球形红细胞增多症(hereditary spherocytosis, HS)。患儿入院后存在转氨酶升高以及胆汁淤积的情况,故给予复方甘草酸苷、双环醇保肝,熊去氧胆酸胶囊口服利胆退黄治疗。向患儿家长交代必要时需行脾切除术。
随访:出院后未予特殊治疗,出院2个月后复查肝功能示天冬氨酸转氨酶30.3 U/L,丙氨酸转氨酶7.4 U/L,γ-谷氨酰胺转移酶9.5 U/L,总胆红素148.8 μmol/L,直接胆红素35.3 μmol/L,间接胆红素113.5 μmol/L。血常规示白细胞5.47×109/L,红细胞3.52×1012/L,血小板301×109/L,血红蛋白101 g/L。现仍继续随访中。
HS典型临床表现为溶血性贫血、黄疸以及肝脾肿大等[1]。实验室检查可见球形红细胞增多、网织红细胞升高、间接胆红素升高、红细胞渗透脆性试验阳性以及Coombs试验阴性等[2]。本例患儿存在黄疸、肝脾肿大、贫血、网织红细胞升高、红细胞渗透脆性试验阳性、Coombs试验阴性,且患儿既往有溶血性贫血、黄疸的病史,故考虑HS的可能。但患儿胆红素以直接胆红素升高为主,考虑胆汁淤积,与HS不符。为明确诊断,本例患儿完善了肝脏病理,结果示胆汁淤积性改变。目前检索到4例HS患者出现胆汁淤积的报道[2,3,4,5]。国内曾有HS患者出现胆汁淤积的报道,考虑与结石导致胆总管梗阻、胆红素生成过多有关,并且由于炎症、缺氧导致肝细胞离子泵或者酶受到影响从而出现[2,3,4]。国外有1例HS合并复发性肝内胆汁淤积2型的患者出现胆汁淤积的报道[5]。本例患儿虽然入院期间以直接胆红素升高为主,肝脏病理提示胆汁淤积,但随访期间患儿直接胆红素可以降至正常,而间接胆红素不能降至正常,且患儿曾有溶血性贫血病史,故考虑本患儿胆汁淤积可能由于受到胆红素生成过多、炎症、缺氧等因素影响,为一过性,而间接胆红素比例应该为持续升高。提示临床遇到胆汁淤积的患儿也需要完善溶血的相关检查,并应动态观察胆红素变化情况。基因检测是确定红细胞膜疾病遗传病因的有效方法[6]。本例患儿入院后进行了基因检测,发现患儿SPTB基因发生了移码突变,从而确诊HS。本例患儿所检测出的c.1791delG,为新发突变,国内外未发现相关报道。目前认为除少数重症患儿,HS患儿的脾切术年龄应大于5岁[5]。本例患儿5岁,且临床贫血较轻,胆红素指标经常规治疗也能明显下降,且脾切除术后并不能有效阻止胆道结石并发症的发生故暂时未予脾切除术治疗。
综上,对于以胆汁淤积起病的患儿,应完善溶血相关检查。对于其中网织红细胞明显升高,且红细胞渗透脆性试验阳性、Coombs试验阴性的患儿,应高度警惕HS的可能,应进一步完善基因检测助诊。本例报道的缺憾:(1)患儿未进行基因血缘分析,故不能明确患儿父母是其生物学意义的父母。(2)因为本例患儿胆汁淤积为一过性,故考虑本例患儿胆汁淤积与基因可能无明显关联,但明确二者有无内在关联,还需要更多病例和基因分析总结。
所有作者均声明不存在利益冲突

























