
本文报道一遗传性凝血因子Ⅶ缺乏症家系及基因分析结果。先证者检测到凝血因子Ⅶ基因发生c.1198 G>C(p.Val400Leu)和IVS6-1 G>A复合杂合突变。前者在中国汉族人群当中尚未见报道。其中c.1198 G>C(p.Val400Leu)突变来自患儿母亲;IVS6-1 G>A来自患儿父亲。IVS6-1 G>A突变为已知致病突变,c.1198 G>C(p.Val400Leu)在100名正常对照人群中未检出该突变。根据美国医学遗传学与基因组学学会(ACMG)遗传变异分类标准与指南,c.1198 G>C突变为可能致病突变。先证者凝血因子Ⅶ基因c.1198 G>C(p.Val400Leu)和IVS6-1 G>A复合杂合突变可能是患儿表现为临床出血症状的遗传学致病因素。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
遗传性凝血因子Ⅶ缺乏症是由于凝血因子Ⅶ基因突变所导致的常染色体隐性出血性疾病,发病率约为1∶500 000。由于凝血因子Ⅶ功能缺陷,影响了机体外源性凝血途径的起始阶段,从而导致临床出血症状。该病在临床上分两种类型:1型表现为凝血因子Ⅶ抗原(FⅦ antigen,FⅦ:Ag)和凝血因子Ⅶ活性(FⅦ activity,FⅦ:C)都降低;2型表现为FⅦ:C降低,但FⅦ:Ag正常[1]。凝血因子FⅦ基因缺陷导致血浆中凝血因子Ⅶ含量减少和(或)功能发生障碍,部分患者可发生出血症状。已报道的凝血因子Ⅶ基因突变类型包括错义或无义突变、剪切位点突变,小的插入、缺失及启动子区域突变等。本文报道一例遗传性凝血因子Ⅶ缺乏症患儿,通过对先证者及其家系成员基因测序,探讨其致病原因。
先证者为男性患儿,4岁,因"反复鼻衄,加重1 d"就诊。患儿出生后有出血部位止血困难病史。入院前1年无明显诱因出现间断鼻出血,量中等,间隔3~5 d。经用云南白药及棉球填塞处理后止血。近期出血间隔时间缩短,出血量多,上述方法不能有效止血。入院前1 d,出血加重,为进一步诊治,收住入院。患儿部分出凝血功能检验指标异常:凝血酶原时间(PT)51.8 s(参考范围:11.0~ 14.0 s,对照值12 s);活化部分凝血活酶时间(APTT)26.8 s(参考范围:25.0~43.0 s,对照值30 s);纤维蛋白原(FIB)2.25 g/L(参考范围:2.00~4.00 g/L);凝血酶时间(TT)17.5 s(参考范围:16.0~18.0 s);凝血因子Ⅶ活性(FⅦ:C)2.8%(参考范围:50.0~150.0%)。患儿父母上述检测指标均在正常参考范围内。患儿为第一胎,父母体健,非近亲结婚。父母双方家族中否认有异常出血患者。母亲孕期无高危因素暴露病史。在患儿父母知情同意的情况下,抽取患儿及其父母乙二胺四乙酸(EDTA)抗凝外周静脉血2~4 ml进行DNA提取。选择本实验室100名健康成人DNA做对照样本。
(1)提取DNA及定量:采用北京天根生化科技有限公司外周血基因组提取试剂盒(TIANGEN TIANamp Genomic DNA Kit)按照说明书提取DNA。TE充分溶解,Nano Drop 2000核酸定量仪测定DNA纯度和浓度。DNA浓度控制在50~250 ng/μl,-20℃保存。(2)PCR扩增及电泳:用在线引物设计软件Primer 3自行设计9对引物以覆盖凝血因子Ⅶ基因全部外显子及其侧翼和启动子区,引物由上海生工生物工程股份有限公司合成。PCR反应条件:94℃变性5 min,然后95℃变性30 s,61℃复性30 s,72℃延伸1 min,34个循环后于72℃再延伸10 min。PCR产物1.5%琼脂糖电泳,切胶纯化后,在ABI 9700 PCR仪上进行测序反应;在ABI 3500DX遗传分析仪上进行毛细管电泳。(3)Sanger测序结果分析:应用序列比对软件SeqMan7.1对Sanger测序结果与凝血因子Ⅶ基因参考序列(NM_019616.3)进行比对分析。
Sanger测序结果与凝血因子Ⅶ基因参考序列(NM_019616.3)经比对发现,患儿存在该基因IVS6-1 G>A、c.1198 G>C(p.Val400Leu)复合杂合突变。患儿父亲携带有凝血因子Ⅶ基因IVS6-1 G>A杂合突变;患儿母亲携带有凝血因子Ⅶ基因c.1198 G>C(p.Val400Leu)杂合突变(图1)。检索人类基因突变数据库(Human Gene Mutation Database,HGMD),凝血因子Ⅶ基因IVS6-1 G>A为已知致病突变。凝血因子Ⅶ基因c.1198 G>C(p.Val400Leu)突变类型国内外尚未见报道,100份对照样本未检出此类突变。
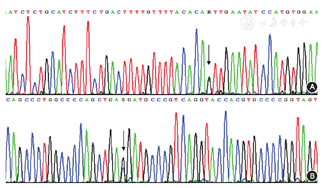
注:A示患者外周血标本检测到父源性致病突变IVS6-1 G>A;B示患者外周血标本检测到母源性突变c.1198 G>C
应用突变危害预测软件PolyPhen-2 v2.2.2r398对凝血因子Ⅶ基因c.1198 G>C突变的致病性进行预测分析,结果为"可能危害"(PROBABLY DAMAGING)得分0.998(敏感性0.18,特异性0.98)。根据ACMG遗传变异分类标准与指南,该突变为可能致病突变。
凝血因子Ⅶ是肝脏合成的维生素K依赖性单链糖蛋白酶原。血浆中,凝血因子Ⅶ主要以酶原形式存在,活性形式的凝血因子Ⅶa仅占1%。成熟凝血因子Ⅶ由406氨基酸残基组成,相对分子质量为45 078。凝血因子Ⅶ基因定位于13号染色体13q34,长度为12 800碱基对,由9个外显子和8个内含子组成。外显子2编码γ-羧基谷氨酸(Gla)区;外显子3编码一个小的疏水区;4、5号外显子编码表皮生长因子样区(EGF);6~8号外显子编码催化区。凝血因子Ⅶ的作用主要是和组织因子(TF)形成复合物(TF·FⅦ)后激活凝血因子X(FX)而启动外源性凝血途径[2]。
凝血因子Ⅶ基因突变是导致凝血因子Ⅶ功能异常,发生临床凝血功能障碍,导致临床出血症状的主要原因。据HGMD(http://www.hgmd.cf.ac.uk/ac/index.php gene=F7)报道,凝血因子Ⅶ基因突变类型包括错义或无义突变、剪切位点突变,小的插入、缺失及启动子区域突变及复杂重排等。其中错义或无义突变约占65%、剪切位点突变约占15%,小的插入、缺失及启动子区域突变占15%,其它突变类型约占5%。凝血因子Ⅶ基因突变与临床表型的关系较为重要,纯合性和复合杂合性基因突变,其凝血因子Ⅶ活性(FⅦ:C)一般在1%~5%之间,多伴有临床症状;而单纯的杂合突变FⅦ:C一般不低于30%,几乎没有什么临床症状,仅有一些特殊的杂合突变可表现为中、轻度的临床出血[3]。本文报道的先证者为凝血因子Ⅶ基因IVS6-1 G>A、c.1198 G>C(p.Val400Leu)复合杂合突变患者。患儿出生后有出血部位止血困难病史。PT 51.8 s(参考范围:11.0~14.0 s,对照值12 s),凝血因子Ⅶ活性(FⅦ:C)2.8%(参考范围:50.0~150.0%)。先证者父母为凝血因子Ⅶ基因IVS6-1 G>A和c.1198 G>C杂合突变携带者,PT、FⅦ:C均在正常参考范围之内,分别为12.1 s和12.7 s、89.3%和91%。先证者父母均无异常出血、出血不止和鼻衄病史,其临床表型与文献报道相符。
有关凝血因子Ⅶ基因IVS6-1 G>A致病突变在中国患者人群中报道较多,可能是中国汉族人的热点突变[4,5]。其分子致病机制可能是由于该突变破坏了GT-AG剪接规则中受体位点的保守序列,使凝血因子Ⅶ基因内含子6不能被正常剪接所致。Yu等[5]对凝血因子Ⅶ基因IVS6-1 G>A突变的致病机制进行体外mRNA转录水平研究。结果表明,突变导致外显子7被跳跃剪接,形成了缺乏7号外显子的异常转录本。因此推测IVS6-1G>A突变后,由于外显子7丢失,从而影响凝血因子Ⅶ蛋白的合成、分泌及生物功能。凝血因子Ⅶ基因c.1198 G>C突变类型,目前国内外文献中尚未见报道。我们对出凝血功能正常的100份健康对照样本进行检测,未发现该突变。应用PolyPhen-2 v2.2.2r398软件对其致病性进行预测分析,PROBABLY DAMAGING得分0.998,为可能致病性突变。根据ACMG遗传变异分类标准与指南,结合患者临床症状,凝血因子Ⅶ基因c.1198 G>C为可能致病性突变。凝血因子Ⅶ基因c.1198 G>C(p.Val400Leu)、IVS6-1 G>A复合杂合突变可能是患者表现为临床出血症状的遗传学致病因素。
遗传性凝血因子Ⅶ缺乏症在临床上以出血症状为主要表现。常见的出血症状为鼻衄、牙龈出血、皮肤瘀点、瘀斑、呕血、黑便及创伤后持续性出血等,严重者发生致命性颅内出血。患者的出血症状与出血程度往往与其FⅦ:C检测水平不一致。丁秋兰等[6]报道了一名由于携带有三种杂合突变的临床出血症患者,其FⅦ:C检测结果仅为1%,而与先证者有相同基因突变的另两位家系成员却无临床症状。Heremann等[3]报道l例凝血因子Ⅶ基因7号内含子短重复序列杂合突变的女性,其FⅦ:C仅为1%,临床表现为月经过多、具有轻度的出血倾向。因此,仅凭FⅦ:C检测无法判断患者病情及愈后。通过基因诊断,可以明确患者基因型,为临床治疗及以后的产前诊断提供帮助。因此,临床遇到FⅦ活性低下的患者时,应当对先证者及其父母凝血因子Ⅶ基因进行Sanger测序,明确是否由于基因突变导致的临床表现。
所有作者均声明不存在利益冲突

























