
探讨神经发育障碍(NDD)患儿的遗传学病因,为其遗传咨询、家庭的再发风险评估及产前诊断提供理论依据。
根据NDD的诊断标准,收集2016年1月至2018年12月来湖南省妇幼保健院小儿康复科就诊的420例NDD患儿,行染色体核型分析和染色体微阵列(CMA)分析。
420例NDD患儿中,14例(3.33%,14/420例)染色体异常,均为全面性发育迟缓/智力障碍(GDD/ID),其中13例通过进一步CMA明确了染色体断裂点位置和缺失或重复的片段范围;共在61例(14.52%,61/420例)患儿中发现致病性拷贝数变异(CNVs)。在这些致病性CNVs中,包括已知的综合征31例(50.82%,30/61例),涉及Angelman/Prader-Will综合征(8例)、Williams综合征(3例)、Phelan-McDermid综合征(3例)等16种综合征以及30例具有临床意义的致病性CNVs。另外还结合荧光原位杂交技术(FISH)诊断了1个因10q26与12p13隐匿性重排引起的智力低下家系。
染色体异常和基因组相关的微缺失重复是NDD患儿重要的遗传学病因,联合应用核型分析、CMA和FISH技术可为NDD患儿提供明确的病因诊断,对患儿的治疗及其父母再生育指导具有重要临床意义。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
儿童神经发育障碍(neurodevelopmental disorders,NDD)是一组病因复杂、严重危害儿童身心健康甚至影响患儿终身的慢性发育性脑功能障碍性疾病。NDD主要分为全面性发育迟缓/智力障碍(global developmental disabilities/intellectual disabilities,GDD/ID)、孤独症谱系障碍(autism spectrum disorders,ASD)、注意缺陷多动障碍(attention deficit hyperactivity disorder,ADHD)等[1],其中不明原因GDD/ID的人群患病率为2%~3%[2],全世界ASD的患病率为1%~2%[3]。NDD给家庭和社会带来沉重的经济和精神负担,严重影响患者和其家人的生活质量,已成为一类全世界高度关注的公共健康问题。
NDD发病机制复杂,其发生既有炎性反应、免疫紊乱及代谢障碍等环境风险因素,又有遗传因素参与。近年来,遗传因素在病因中构成日渐突出,主要包括染色体异常、基因/基因组变异。据统计,遗传因素中染色体数目和结构异常占GDD/ID人群的25%~30%[4]。大规模研究数据表明,拷贝数变异(copy number variation,CNV)与NDD的发病密切相关[5,6]。回顾性研究结果显示,NDD患儿的染色体微阵列分析(chromosome microarray analysis,CMA)检测结果,可为其临床治疗管理提供有效依据[7]。基于多个中心CMA结果的积累,欧美国家和中华医学会儿科学分会内分泌遗传代谢学组先后发布共识,建议将CMA作为不明原因发育迟缓(DD)/ID、ASD等临床表型的一线检测手段[8]。
本研究应用核型分析技术和CMA技术对420例NDD患儿进行染色体和全基因组CNVs分析,明确其遗传学病因,探讨NDD患儿相关CNVs与表型的关联,为疾病的预防及治疗提供理论依据。
选取2016年1月至2018年12月在湖南省妇幼保健院小儿康复科就诊的GDD/ID、ASD和ADHD患儿。共纳入患儿420例,其中男278例,女142例;年龄4个月~16岁,其中GDD/ID患儿259例,ASD患儿155例,ADHD患儿6例。诊断标准:(1)GDD:婴幼儿采用Gesell发育量表测评发育商(DQ)<75分;(2)ID:学龄前儿童和儿童使用韦氏智力测试(IQ)<70分;(3)ASD:符合美国精神障碍诊断统计手册第5版(DSM-Ⅴ)中诊断标准[9];(4)ADHD:符合美国精神障碍诊断统计手册第4版(DSM-Ⅳ)中诊断标准[10]。排除标准:(1)母亲孕期营养不良、毒性药物接触史、缺氧、围生期感染、外伤;(2)已明确遗传病因的GDD/ID,如21-三体、18-三体、脆性X综合征及苯丙酮尿症等。对于CMA发现CNVs的部分患儿,再次抽取其父母外周血进一步检测比对。本研究通过医院医学伦理委员会审批(批准文号:2019-S034),并获得患儿监护人的同意,且签署知情同意书。
抽取患儿外周血2 mL,肝素钠抗凝,按照常规外周血染色体制备方法进行细胞培养、收获及制片,G显带,核型分析。核型异常,加大计数至50个分裂以上,每种类型分析3~5个分裂相。
采集患儿静脉血2 mL,应用QIAGEN试剂盒(德国QIAGEN公司)提取基因组DNA。应用美国Affymetrix公司CytoScan750k芯片对样本DNA进行检测,按照相关试剂说明书进行操作。对所得原始数据应用Affymetrix Chromosome Analysis Suite Software进行分析。
将检出的CNVs结果参照CNVs多态性数据库(DGV)、人类异常表型数据库(DECIPHER)以及与疾病相关的人类基因组变异数据库(Clin Var)、在线孟德尔遗传疾病数据库(OMIM)、湖南省妇幼保健院数据库和统一的中国人群CNV数据库等数据库及相关文献。按照美国医学遗传学与基因组学学会(ACMG)指南[11],将CNVs分为5个等级:(1)致病性CNVs;(2)可能致病性CNVs;(3)意义不明的CNVs(VOUS);(4)可能为良性CNVs;(5)良性CNVs。对于临床意义不明CNVs,需验证该CNVs是新生变异还是遗传自父母,再进行判断。
为进一步诊断1例检测出缺失并重复CNV患儿的病因,应用美国Abbott-Vysis公司的10pter(绿色)/10qter(红色)、12pter(绿色)/12qter(红色)的特异性探针对患儿及父母外周血中期分裂进行FISH杂交,扫片及计数。
420例NDD患儿中14例(3.33%)存在染色体异常,均为GDD/ID患儿,包括8例(病例1、2、4、5、7、8、10、12)染色体末端缺失,4例(病例3、9、13、14)染色体中间区域的缺失或重复,1例(病例11)携带不明来源的标记染色体(supernumerary marker chromosome,SMC),还有1例(病例6)为相互易位;病例11通过染色体核型分析和CMA检测,诊断为PKS。检测结果见表1。
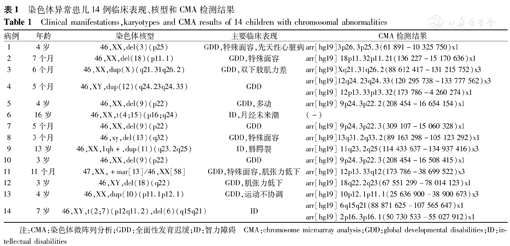
染色体异常患儿14例临床表现、核型和CMA检测结果
Clinical manifestations,karyotypes and CMA results of 14 children with chromosomal abnormalities
染色体异常患儿14例临床表现、核型和CMA检测结果
Clinical manifestations,karyotypes and CMA results of 14 children with chromosomal abnormalities
| 病例 | 年龄 | 染色体核型 | 主要临床表现 | CMA检测结果 |
|---|---|---|---|---|
| 1 | 4岁 | 46,XX,del(3)(p25) | GDD,特殊面容,先天性心脏病 | arr[hg19]3p26.3p25.3(61 891-10 325 750)x1 |
| 2 | 7个月 | 46,XX,del(18)(p11.1) | GDD,特殊面容 | arr[hg19] 18p11.32p11.21(136 227-15 170 636)x1 |
| 3 | 6个月 | 46,XX,dup(X)(q21.31q26.2) | GDD,双下肢肌力差 | arr[hg19]Xq21.31q26.2(88 612 417-131 215 752)x3 |
| 4 | 5个月 | 46,XY,dup(12)(q24.23q24.33) | GDD | arr[hg19]12q24.23q24.33(120 295 738-133 777 562)x3 |
| arr[hg19] 12p13.33p13.32(173 786-4 260 274)x1 | ||||
| 5 | 4岁 | 46,XX,del(9)(p22) | GDD,多动 | arr[hg19] 9p24.3p22.2(208 454-16 654 154)x1 |
| 6 | 16岁 | 46,XX,t(4;15)(p16;q24) | ID,月经未来潮 | (-) |
| 7 | 5个月 | 46,XX,del(9)(p22) | GDD | arr[hg19] 9p24.3p22.3(309 107-15 060 328)x1 |
| 8 | 3个月 | 46,xy,del(13)(q32) | GDD,特殊面容 | arr[hg19]13q31.2q33.2(89 163 298-105 123 292)x1 |
| 9 | 13岁 | 46,XX,1qh+,dup(11)(q23.2q25) | ID,唇腭裂 | arr[hg19] 11q23.2q25(114 433 637-134 937 416)x3 |
| 10 | 3岁 | 46,XX,del(9)(p22) | GDD | arr[hg19] 9p24.3p22.3(208 454-16 508 415)x1 |
| 11 | 11个月 | 47,XX,+mar[13]/46,XX[58] | GDD,特殊面容,肌张力低下 | arr[hg19] 12p13.33q12(173 786-38 699 522)x3 |
| 12 | 3岁 | 46,XY,del(18)(q22) | GDD,肌张力低下 | arr[hg19] 18q22.2q23(67 551 299-78 014 123)x1 |
| 13 | 4岁 | 46,XX,dup(10)(p11.1p12.1) | GDD,运动不协调 | arr[hg19] 10p12.1p11.1(25 636 900-38 900 673)x3 |
| 14 | 7岁 | 46,XY,t(2;7)(p12q11.2),del(6)(q15q21) | ID | arr[hg19] 6q15q21(88 871 625-107 565 647)x1 |
| arr[hg19] 2p16.3p16.1(50 730 533-55 027 912)x1 |
注:CMA:染色体微阵列分析;GDD:全面性发育迟缓;ID:智力障碍 CMA:chromosome microarray analysis;GDD:global developmental disabilities;ID:intellectual disabilities
420例患儿均进行CMA检测,其中61例(14.52%)(包括染色体异常患儿)发现致病性CNVs,GDD/ID、ASD及ADHD患儿的检出率分别为19.69%(51/259例)、6.45%(10/155例)、0(0);17例(4.05%,17/420例)发现可能致病性CNVs。其中30例患儿发现VOUS,进一步对其父母进行CMA检测,结果显示9例遗传自表型正常父母的一方,被重新判定为可能为良性,因此本研究中共21例(5.00%,21/420例)患儿发现VOUS。321例(76.43%,321/420例)患儿的CNVs为可能良性和良性,结果见表2。
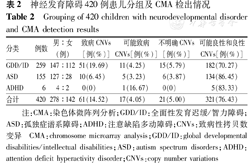
神经发育障碍420例患儿分组及CMA检出情况
Grouping of 420 children with neurodevelopmental disorder and CMA detection results
神经发育障碍420例患儿分组及CMA检出情况
Grouping of 420 children with neurodevelopmental disorder and CMA detection results
| 分类 | 例数 | 男:女(例) | 致病CNVs[例(%)] | 可能致病CNVs[例(%)] | 不明确CNVs[例(%)] | 可能良性和良性CNVs[例(%)] |
|---|---|---|---|---|---|---|
| GDD/ID | 259 | 147:112 | 51(19.69) | 11(4.25) | 15(5.79) | 182(70.27) |
| ASD | 155 | 127:28 | 10(6.45) | 5(3.23) | 6(3.87) | 134(86.45) |
| ADHD | 6 | 4:2 | 0(0) | 1(16.67) | 0(0) | 5(83.33) |
| 合计 | 420 | 278:142 | 61(14.52) | 17(4.05) | 21(5.00) | 321(76.43) |
注:CMA:染色体微阵列分析;GDD/ID:全面性发育迟缓/智力障碍;ASD:孤独症谱系障碍;ADHD:注意缺陷多动障碍;CNVs:致病性拷贝数变异 CMA:chromosome microarray analysis;GDD/ID:global developmental disabilities/intellectual disabilities;ASD:autism spectrum disorders;ADHD:attention deficit hyperactivity disorder;CNVs:copy number variations
61例致病性CNVs中,31例患儿涉及16种综合征(50.82%,31/61例),包括8例Angelman/Prader-Will综合征、3例Williams综合征、3例Phelan-McDermid综合征、2例5q14.3微缺失综合征、2例Smith-Magenis综合征、1例1p36微缺失综合征、1例Mowat-Wilson综合征、1例2q33.1微缺失综合征、1例Miller-Dieker综合征、1例DiGeorge综合征、3例9p缺失综合征、1例3p缺失综合征、1例Pallister-Killian综合征(Pallister-Killian syndrome,PKS)、1例13q缺失综合征、1例18p缺失综合征、1例18q缺失综合征,结果见表3。
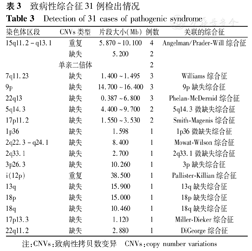
致病性综合征31例检出情况
Detection of 31 cases of pathogenic syndrome
致病性综合征31例检出情况
Detection of 31 cases of pathogenic syndrome
| 染色体区段 | CNVs类型 | 片段大小(Mb) | 例数 | 关联的综合征 |
|---|---|---|---|---|
| 15q11.2-q13.1 | 重复 | 5.870~10.100 | 4 | Angelman/Prader-Will综合征 |
| 缺失 | 5.200 | 2 | ||
| 单亲二倍体 | 2 | |||
| 7q11.23 | 缺失 | 1.400~1.495 | 3 | Williams综合征 |
| 9p | 缺失 | 14.700~16.400 | 3 | 9p缺失综合征 |
| 22q13 | 缺失 | 0.387~6.800 | 3 | Phelan-McDermid综合征 |
| 5q14.3 | 缺失 | 4.400~9.700 | 2 | 5q14.3微缺失综合征 |
| 17p11.2 | 缺失 | 1.550~3.530 | 2 | Smith-Magenis综合征 |
| 1p36 | 缺失 | 1.598 | 1 | 1p36微缺失综合征 |
| 2q22.3-q24.1 | 缺失 | 8.400 | 1 | Mowat-Wilson综合征 |
| 2q33.1 | 缺失 | 2.700 | 1 | 2q33.1微缺失综合征 |
| 3p26.3 | 缺失 | 10.260 | 1 | 3p缺失综合征 |
| i(12p) | 重复 | 38.500 | 1 | Pallister-Killian综合征 |
| 13q | 缺失 | 15.900 | 1 | 13q缺失综合征 |
| 18p | 缺失 | 15.000 | 1 | 18p缺失综合征 |
| 18q | 缺失 | 10.460 | 1 | 18q缺失综合征 |
| 17p13.3 | 缺失 | 1.120 | 1 | Miller-Dieker综合征 |
| 22q11.2 | 缺失 | 2.880 | 1 | DiGeorge综合征 |
注:CNVs:致病性拷贝数变异 CNVs:copy number variations
另外还检出30例具有临床意义的致病性CNVs,其中13例为缺失型,15例为重复型,2例为缺失并重复型。其中1例缺失并重复型患儿(病例70),女,15岁,因"出生后智力发育明显低于同龄人"就诊,但生活可自理,一直未予重视;从小体弱,存在喂养困难史,14岁前有遗尿现象,眼睛斜视后经手术矫正,上腭高尖,IQ为60;CNVs提示患儿10q26.3存在约4 Mb缺失,且12p13.33存在约899 kb重复,FISH结果提示该患儿致病性CNVs源于父亲隐匿性染色体重排,并在产前诊断中诊断出该家系一类似胎儿,结果见表4和图1,图2,图3。


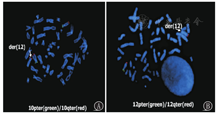

染色体缺失并重复型患儿(病例70)检测情况
Detection of a microdeletion and microrepetition patient (case 70)
染色体缺失并重复型患儿(病例70)检测情况
Detection of a microdeletion and microrepetition patient (case 70)
| 项目 | 普通G显带核型分析 | CMA检测结果 | FISH检测结果 |
|---|---|---|---|
| 患儿 | 46,XX | 10q26.3存在约4 Mb缺失,12p13.33存在约899 kb重复 | ish der(10)t(10;12)(q26;p13)(10pter+,10qter-,12pter+)pat |
| 父亲 | 46,XY | / | ish der(10)t(10;12)(q26;p13)(10pter+,10qter-,12pter+),der(12)t(10;12)(q26;p13)(12pter-,10qter+,12qter+) |
| 母亲 | 46,XX | / | 未发现异常 |
| 家系类似胎儿 | 46,XY | 10q26.3存在4.6 Mb重复,12p13.33存在899 kb缺失 | ish der(12)t(10;12)(q26;p13)( 12pter-,10qter+,12qter+)pat |
注:CMA:染色体微阵列分析;FISH:荧光原位杂交技术 CMA:chromosome microarray analysis;FISH:fluorescence in situ hybridization
NDD是一类临床表型异质性强、病因复杂的疾病,随着分子细胞检测技术的快速发展,已证实了数百基因异常或染色体异常可导致NDD的发生[12,13]。Miller等[11]分析GDD/ID、多发畸形和ASD患者,结果显示G显带核型分析技术的检出率为3%,CMA的检出率为15%~ 20%。国内外数据报告,10%~19%的GDD/ID患者可通过CMA找到病因[14,15]。Shen等[16]对848例ASD患儿进行CMA检测,结果显示致病性CNVs的检出率为7%。本研究对420例NDD患儿进行染色体核型分析和CMA分析,发现14例(3.33%)染色体异常;61例(14.52%)检测到致病性CNVs,其中在GDD/ID和ASD患儿中的检出率分别为19.69%、6.45%,这与之前报道[14,15,16]的数据基本相符。6例ADHD均未在染色体和CMA水平上寻找到致病原因,这可能是由于纳入本研究的病例样本数量过少所导致,ADHD患儿遗传病因学研究还有待在大样本中展开。
传统G显带核型分析可以发现染色数目异常及5~10 Mb以上的结构变异。本研究中发现了14例染色体异常,均为GDD/ID患儿。进一步利用CMA定位了8例染色体末端缺失片段的断裂点;明确了4例染色体中间区域的缺失或重复的片段范围;确定了病例11中SMC为i(12p),病例6为平衡相互易位。病例11通过染色体核型分析和CMA检测,结合临床表型诊断为PKS。PKS中的i(12p)在不同组织中嵌合比例不同,异常核型常存在于皮肤成纤维细胞中,而外周血淋巴细胞呈低比例嵌合或缺失,嵌合比例随年龄增加逐渐下降,由于异常的染色体在有丝分裂过程中不稳定,在培养时易丢失,造成外周血淋巴细胞染色体核型分析敏感性低。CMA是直接提取DNA进行检测,较大程度上避免了体外培养过程中的丢失,提高了PKS诊断的敏感性和准确性。病例6为染色体平衡易位患儿,但CMA未发现致病性CNVs,其ID的致病原因可能是由于其他基因突变,这需要通过进一步高通量测序来寻找。CMA分辨率高,但无法检测平衡易位、倒位和复杂重排。将染色体核型分析与CMA相结合,可准确分析不平衡易位的位置、片段大小和来源,对于评估患儿基因型与临床表型的相关性具有重要意义。
检测出致病性CNVs的61例患儿中,31例(50.82%)为已知的16种综合征,其中检出率最高的是Angelman/Prader-Will综合征(8例),其次为Williams综合征(3例)、Phelan-McDermid综合征(3例)和Smith-Magenis综合征(3例),这与已报道的NDD致病热点区域类似[7] 。此外还检测出Mowat-Wilson综合征、2q33.1微缺失综合征等罕见微缺失微重复综合征以及9p缺失综合征、3p缺失综合征等6种综合征。2q33.1微缺失综合征是由2q33.1区域杂合性缺失或关键基因突变而引起的一种临床综合征,SATB2基因为致病基因,该基因缺失突变会导致GDD/ID和言语发育缺失或受限,颅面畸形,包括小颌畸形、睑裂下垂、腭裂等[17]。本研究中病例36,女,有先天性上唇腭裂手术史,2岁时不能爬行和站立,语言落后,CMA发现2q33.1存在约2.700 Mb的缺失,包含了SATB2基因,且表型与以往报道[18]的基本相符,因此被诊断为2q33.1微缺失综合征。
本研究还检出30例具有临床意义的致病性CNVs,包括缺失型、重复型、缺失并重复型3种类型,丰富了致病突变数据库,为新的综合征发现提供了线索。本研究中,病例70的致病性CNVs为缺失并重复型,该患儿外周血普通G显带染色体未见明显异常,CMA结果提示10q26.3存在约4 Mb缺失,12p13.33存在约899 kb重复,DECIPHER数据库报道上述区间内缺失可导致严重智力障碍和斜视,这与患儿的表型相符。进一步验证患儿父母,患儿父母染色体未见异常,FISH检测提示其父亲核型为隐匿性相互易位t(10;12)(q26;p13)的携带者。患儿遗传了父亲其中一条衍生的10号染色体,导致12p部分三体和10q部分单体,从而出现了异常表型。当患儿母亲再次妊娠时,抽取羊水进行CMA检测,发现10q26.3存在4.6 Mb重复,12p13.33存在899 kb缺失,DECIPHER数据库提示这将导致自闭症和系统性发育迟缓、肌张力低下等发育异常;FISH验证结果显示胎儿遗传了父亲一条由t(10;12)形成的der(12)衍生染色体,导致了10p部分三体和12q部分单体,最终选择终止妊娠避免类似患儿的出生。该病例提示,当CNVs提示染色体同时出现缺失和重复时,需要验证父母是否为平衡易位携带者,为同胞的再发风险评估及产前诊断提供重要依据。
此外,本研究还在21例患儿中检出VOUS,这给解释患者表型带来了困难与挑战。尽管目前判断它们是致病变异还是良性变异的证据还不足,但随着人类基因组中的CNVs陆续被发现以及相关数据库的不断完善,这些VOUS可能对某些复杂疾病的致病原因具有潜在提示作用,或为今后的分析提供拷贝数多态性的参考。
综上,本研究联合核型分析、CMA、FISH等3种技术,对420例NND患儿进行检测分析,在61例患者中检出了致病性CNVs,为这部分患者的诊断、康复治疗及预后评估提供了依据,同时为其家庭再生育时患病风险的评估提供了理论基础。
所有作者均声明不存在利益冲突

























