
心肌病是一组异质性心肌疾病,可由各种原因(常为遗传因素)引起,能够导致心力衰竭、心律失常和猝死。原发性心肌病包括遗传性肥厚型心肌病、致心律失常右室心肌病、线粒体心肌病、混合性(遗传性及获得性)扩张型心肌病和限制型心肌病、左心室致密化不全以及其他未分类的心肌病。借助基因组学技术,在人群中发现的一些常见突变与疾病的关联已被鉴定。这些突变的体内和体外功能研究为疾病的发生机制和治疗提供了有用的线索。本指南在参考国内外本领域的基础研究、临床研究和其他国家的相关指南共识的基础上,对不同类型遗传型心肌病的表型、诊断、治疗及遗传咨询进行了总结,期望有助于患者临床管理的规范化。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
心肌病作为一组影响心脏功能的疾病,可由多种原因(常为遗传因素)引起,能够导致心力衰竭、心律失常和猝死。世界卫生组织/国际学会和心脏病联合会工作组1995年建议将心肌病分为心肌病和原发性两类[1]。美国心脏协会(American Heart Association,AHA)2006年[2]又进行了进一步的完善,其中特异性心肌病包括与心肌炎、特定心脏疾病或全身疾病相关的心肌疾病,而原发性心肌病则是一组以原发性心肌结构和功能异常为特征的疾病,在病理生理学上又分为遗传性肥厚型心肌病(hypertrophic cardiomyopathy,HCM)、致心律失常右室心肌病(arrhythmogenic right ventricular cardiomyopathy,ARVC)、线粒体心肌病(mitochondrial cardiomyopathy,MCM)、混合性(遗传性及获得性)的扩张型心肌病(dilated cardiomyopathy,DCM)和限制型心肌病(restrictive cardiomyopathy,RCM)、左心室致密化不全(left ventricular noncompaction cardiomyopathy,LVNC)以及其他未分类的心肌病。其中LVNC被AHA归类为遗传性心肌病[2],而被欧洲心脏病协会定义为未分类实体[3]。患者超声心动图所示的LVNC同时与扩张型心肌病、肥厚型心肌病、限制型心肌病存在关联,因此在使用该术语时需谨慎。LVNC仅描述心肌病的形态而非功能特征,对不同类型的心肌病进行诊断时应做好甄别。2019年,心律协会专家发表了评估心律失常心肌病的共识,提出用推荐等级和证据级别系统组成的评分系统,借助对益处与风险的权衡(采用Ⅰ、Ⅱa、Ⅱb和Ⅲ分类)以及证据等级评分(A、B-R、B-NR、C-LD、C-EO)来表示证据的质量和数量[4]。
HCM通常被定义为不明原因所致的左心室肥厚(left ventricular hypertrophy,LVH),排除了其他心脏或全身性疾病的影响,例如压力超负荷(长期高血压、主动脉狭窄)等。1958年Teare[5]从形态学角度描述了首例HCM。HCM的临床表现多样,从完全无症状到舒张功能障碍、流出道梗阻、进行性心力衰竭、各种快速性心律失常和心源性猝死等。常见的症状包括呼吸困难(尤其在运动时)、胸痛、心悸、晕厥前期和晕厥。
ARVC最早在1982年[6]被描述,表现为心肌逐渐被纤维脂质替代,主要累及右心室,也可累及左心室[4,7,8],易于发生室性心动过速,是年轻人和运动员猝死的原因之一。ARVC心肌病变常发生于右心室尖部、流入道和流出道。典型的ARVC相关心律失常为右室起源呈左束支阻滞图形的室性心动过速,常见的症状包括心悸、晕厥和猝死。
MCM 1998年由Marin-Garcia等[9]提出并命名,其特征为线粒体呼吸链原发功能障碍导致ATP缺乏。其临床表型多样,具有HCM、扩张型心肌病、LVNC等的临床特征,并伴有各种心律失常。
DCM又分为获得型、遗传型和混合型。获得型DCM常见的原因是缺血性损伤(如冠状动脉疾病、心肌梗死)、感染、免疫等;遗传性DCM主要由基因突变引起(表1),而混合型DCM则兼具后天和遗传因素。DCM患者早期可无症状,几年后出现心输出量减少(疲劳、劳累时呼吸困难)、体循环/肺循环淤血(阵发性夜间呼吸困难、端坐呼吸、水肿)等心衰症状,常伴有心律失常和/或传导系统疾病,晚期可发生左心室附壁血栓,导致体循环栓塞。
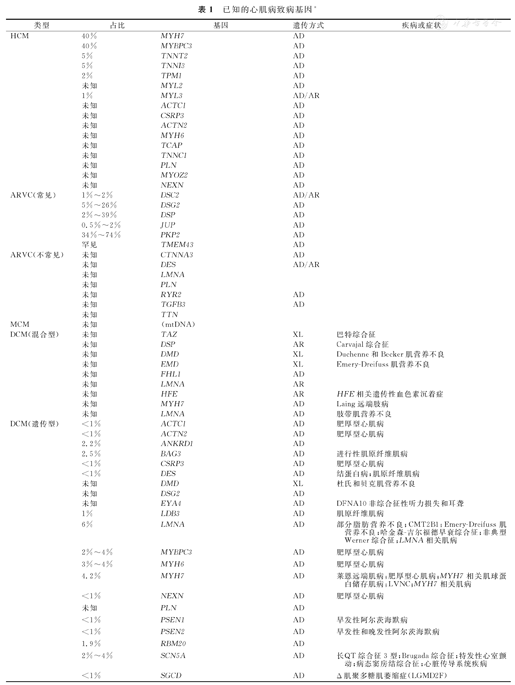
已知的心肌病致病基因*
已知的心肌病致病基因*
| 类型 | 占比 | 基因 | 遗传方式 | 疾病或症状 |
|---|---|---|---|---|
| HCM | 40% | MYH7 | AD | |
| 40% | MYBPC3 | AD | ||
| 5% | TNNT2 | AD | ||
| 5% | TNNI3 | AD | ||
| 2% | TPM1 | AD | ||
| 未知 | MYL2 | AD | ||
| 1% | MYL3 | AD/AR | ||
| 未知 | ACTC1 | AD | ||
| 未知 | CSRP3 | AD | ||
| 未知 | ACTN2 | AD | ||
| 未知 | MYH6 | AD | ||
| 未知 | TCAP | AD | ||
| 未知 | TNNC1 | AD | ||
| 未知 | PLN | AD | ||
| 未知 | MYOZ2 | AD | ||
| 未知 | NEXN | AD | ||
| ARVC(常见) | 1%~2% | DSC2 | AD/AR | |
| 5%~26% | DSG2 | AD | ||
| 2%~39% | DSP | AD | ||
| 0.5%~2% | JUP | AD | ||
| 34%~74% | PKP2 | AD | ||
| 罕见 | TMEM43 | AD | ||
| ARVC(不常见) | 未知 | CTNNA3 | AD | |
| 未知 | DES | AD/AR | ||
| 未知 | LMNA | |||
| 未知 | PLN | |||
| 未知 | RYR2 | AD | ||
| 未知 | TGFB3 | AD | ||
| 未知 | TTN | |||
| MCM | 未知 | (mtDNA) | ||
| DCM(混合型) | 未知 | TAZ | XL | 巴特综合征 |
| 未知 | DSP | AR | Carvajal综合征 | |
| 未知 | DMD | XL | Duchenne和Becker肌营养不良 | |
| 未知 | EMD | XL | Emery-Dreifuss肌营养不良 | |
| 未知 | FHL1 | AD | ||
| 未知 | LMNA | AR | ||
| 未知 | HFE | AR | HFE相关遗传性血色素沉着症 | |
| 未知 | MYH7 | AD | Laing远端肢病 | |
| 未知 | LMNA | AD | 肢带肌营养不良 | |
| DCM(遗传型) | <1% | ACTC1 | AD | 肥厚型心肌病 |
| <1% | ACTN2 | AD | 肥厚型心肌病 | |
| 2.2% | ANKRD1 | AD | ||
| 2.5% | BAG3 | AD | 进行性肌原纤维肌病 | |
| <1% | CSRP3 | AD | 肥厚型心肌病 | |
| <1% | DES | AD | 结蛋白病;肌原纤维肌病 | |
| 未知 | DMD | XL | 杜氏和贝克肌营养不良 | |
| 未知 | DSG2 | AD | ||
| 未知 | EYA4 | AD | DFNA10非综合征性听力损失和耳聋 | |
| 1% | LDB3 | AD | 肌原纤维肌病 | |
| 6% | LMNA | AD | 部分脂肪营养不良;CMT2B1;Emery-Dreifuss肌营养不良;哈金森-吉尔福德早衰综合征;非典型Werner综合征;LMNA相关肌病 | |
| 2%~4% | MYBPC3 | AD | 肥厚型心肌病 | |
| 3%~4% | MYH6 | AD | 肥厚型心肌病 | |
| 4.2% | MYH7 | AD | 莱恩远端肌病;肥厚型心肌病;MYH7相关肌球蛋白储存肌病;LVNC;MYH7相关肌病 | |
| <1% | NEXN | AD | 肥厚型心肌病 | |
| 未知 | PLN | AD | ||
| <1% | PSEN1 | AD | 早发性阿尔茨海默病 | |
| <1% | PSEN2 | AD | 早发性和晚发性阿尔茨海默病 | |
| 1.9% | RBM20 | AD | ||
| 2%~4% | SCN5A | AD | 长QT综合征3型;Brugada综合征;特发性心室颤动;病态窦房结综合征;心脏传导系统疾病 | |
| <1% | SGCD | AD | Δ肌聚多糖肌萎缩症(LGMD2F) | |
| 未知 | TAZ | XL | 巴特综合征;2型心内膜弹力纤维增生症;家族性孤立性LVNC | |
| 1% | TCAP | AD | FHC;LGMD2G | |
| 1.1% | TMPO | AD | ||
| <1%~1.3% | TNNC1 | AD | 肥厚型心肌病 | |
| 1.3% | TNNI3 | AD | 肥厚型心肌病(AD);限制型心肌病(AD) | |
| <1% | TNNI3 | AR | ||
| 2.9% | TNNT2 | AD | 肥厚型心肌病;LVNC;TNNT2相关家族性限制型心肌病 | |
| <1%~1.9% | TPM1 | AD | 肥厚型心肌病 | |
| 10%~20% | TTN | AD | LGMD2J;肌病,早发,伴有致死心肌病;肌病,近端,早期呼吸肌受累;胫骨肌营养不良,迟发性 | |
| 1% | VCL | AD | ||
| RCM | 未知 | TTN | AR | |
| RCM(浸润型) | 未知 | TTR | 淀粉样变性 | |
| 未知 | AGXT、GRHPR、HOGA1 | AR | 高草酸尿症 | |
| 未知 | GLA | XL | Fabry病 | |
| RCM(贮积病) | 未知 | GBA | AR | Gaucher病 |
| 未知 | HAMP、HFE、HFE2、HJV、PNPLA3、SLC40A1、TfR2 | AR | 遗传性血色素沉着症 | |
| 未知 | IDUA | AR | 粘多糖贮积症Ⅰ型(Hurler综合征) | |
| 未知 | IDS | XLR | 粘多糖贮积症Ⅱ型(Hunter综合征) | |
| 未知 | NPC1、NPC2、SMPD1 | AR | Niemann-Pick病 | |
| RCM(非浸润型) | 未知 | BAG3、CRYAB、DES、DNAJB6、FHL1、FLNC、LDB3、MYOT | AD | 肌原纤维肌病 |
| 未知 | ABCC6 | 假黄瘤弹性体 | ||
| 未知 | ATCT、β-MHC、TNNT2、TNNI3、TNNC1、DES、MYH、MYL3、CRYAB | 肌节蛋白病 | ||
| 未知 | WRN | Werner综合征 | ||
| RCM(心肌内) | 未知 | BMP5、BMP7、TAZ | 心内膜纤维化 | |
| LVNC1 | DTNA | |||
| LVNC3 | LDB3 | |||
| LVNC4 | ACTC1 | |||
| LVNC5 | MYH7 | |||
| LVNC6 | TNNT2 | |||
| LVNC7 | MIB1 | |||
| LVNC8 | PRDM16 | |||
| LVNC9 | TPM1 | |||
| LVNC10 | MYBPC3 | |||
| LVNC2 | 11p15 |
数据来自在线资源(GeneReviews等);AD:常显遗传;AR:常隐遗传;XL:X连锁遗传;XLR:X连锁隐性遗传
RCM以心室腔容积正常或缩小、双心房扩大为特征。心肌僵硬度增加导致心室充盈受损,伴有限制性生理学改变,但收缩功能正常。左心室壁厚度和瓣膜通常正常,临床表现为乏力、耐力下降和呼吸困难,严重者还会出现水肿、端坐呼吸、肝肿大、少尿、腹水及消化道淤血等心衰特征,可伴有各种心律失常。
LVNC是一种遗传性疾病,其特征为左心室内出现过多和异常的小梁,可能的原因是心脏的发育在终末阶段发生停滞,无法完全形成致密的心肌[2]。左心室顶端通常呈海绵状,中外侧和下方出现小梁异常。部分可影响右心室,造成右心室或双心室致密化不全。左心室心肌由致密层和非致密层构成,伴有突出的小梁和小梁间的深凹[10,11]。LVNC的临床表现包括左心室收缩障碍、室性心律失常以及与运动相关的胸痛和心悸,其特征高度可变,从无症状到严重心衰甚至猝死。常见者有室性心动过速和房颤。LVNC的表型范围非常广,可分为良性(左心室大小、厚度、收缩/舒张功能正常,无早发性心律失常)、右心室、双心室、DCM、HCM、RCM、混合式(HCM和DCM或DCM和RCM组合)、先天性心脏病和致心律失常等9种形式[12,13]。
作为一种常染色体显性(常显)遗传病,HCM在美国的患病率为1/500[14],在中国为1/1250[15],且具有与年龄相关的差异表达率和外显率。HCM的表型贯穿于从婴儿到老年的整个生命阶段。ARVC的人群患病率为1/1250~1/1000[16],在某些地区更高,在意大利和希腊(纳克索斯岛)甚至高达0.4%~0.8%[17]。MCM呈母系遗传。因群体遗传学瓶颈和建立者效应,MCM的流行病学数据存在争议。约2%的携带mtDNA A3243G突变者被诊断有心肌病,而线粒体病患儿患心肌病的比例则高达40%[18]。DCM的人群发病率约为1/18 182,人群患病率为1/2778[19],在后期具有高死亡率,是心脏移植的常见指征。RCM的人群发病率相对要低很多,但预后很差。Malčić)等[20]曾在121例诊断为心肌病的儿童和青少年中发现6例RCM,占4.8%。LVNC在婴儿中的发病率为0.81/100 000,在儿童中为0.12/100 000[21],在成年人中为1.4/10 000[22]。
























