
探讨遗传性球型红细胞增多症(HS)的临床特点及相关基因突变。
选择2017年1月1日至2019年7月31日,于昆明市儿童医院门诊及住院诊断为HS的10例患儿为研究对象。其中,女性患儿为3例,男性为7例,平均年龄为4.5岁。收集这10例HS患儿一般临床资料,采用高通量测序技术对其进行HS相关基因突变检测。回顾性分析HS患儿的临床特征、相关基因突变及疗效。本研究遵循的程序符合2013年修订版《世界医学协会赫尔辛基宣言》的要求。
①本研究10例患儿均以贫血、黄疸、脾大为临床表现。贫血以小细胞性贫血为主,重度贫血为4例,中度贫血为4例,轻度贫血为2例。患儿脾大以轻、中度为主,轻度脾大为4例,中度脾大为6例,无一例为重度脾大。患儿输血次数不一。3例患儿外周血球形红细胞比例增多,其余7例正常。4例红细胞渗透脆性增加,其余6例正常。患儿地中海贫血基因、铁蛋白及红细胞葡萄糖-6-磷酸脱氢酶(G-6-PD)水平均正常。②高通量基因测序结果显示,10例患儿中,6例存在ANK1、2例存在SPTA1、1例存在SPTB基因突变,1例存在SPTB基因2~3号外显子疑似重复。其中,ANK1基因c.4585C>T(p.R1529X)、c.3877C>T(p.R1293X)突变在人类基因突变数据库(HGMD)专业版中,已有文献报道与球形红细胞增多症相关。ANK1基因c.1471C>T(p.Q491X)、c.1817delT(p.L606Cfs*31)、c.4390+5G>T(IVS36dsG-T+5)、c.4189C>T(p.L1397F)突变,在HGMD专业版中未见报道。根据美国医学遗传学与基因组学学会(ACMG)指南,考虑ANK1基因c.1471C>T(p.Q491X)、c.1817delT(p.L606Cfs*31)突变为新发可疑致病突变。SPTA1基因c.4766G>A(p.W1589X)、c.5432G>A(p.R1811Q)突变,SPTB基因2~3号外显子疑似重复未见文献报道。其中,考虑SPTA1基因c.4766G>A(p.W1589X)突变为新发可疑致病突变。SPTB基因c.4735C>T(p.R1579X)突变已有文献报道。SPTB基因c.797_798del(p.I266Nfs*28)未见文献报道,并且考虑为新发可疑致病突变。③本研究4例接受部分脾动脉栓塞术的患儿中,2例术后未再接受输血治疗;1例术后6个月内,未再接受输血治疗,6个月后每2~3个月接受1次输血治疗,1例失访。其余6例未接受部分脾动脉栓塞术的患儿中,1例每2个月接受输血治疗1次;5例血红蛋白(Hb)值>90 g/L。
对于临床特征不典型的患者可以采用高通量测序技术辅助诊断HS。本研究发现4个未见文献报道的ANK1及SPTA、SPTB基因突变。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
遗传性球型红细胞增多症(hereditary spherocytosis,HS)为一种遗传性溶血性疾病,其临床特征为不同程度的溶血性贫血、黄疸、脾大,外周血球型红细胞增多。HS在任何年龄人群均可发病,以婴儿与儿童多见,男、女性均可发病。HS是由于多种红细胞膜蛋白基因异常,导致膜蛋白的质量与数量异常所致。HS患者中约70%为显性遗传,25%为隐性遗传,5%为新发突变[1]。HS的致病基因包括ANK1、SLC4A1、STPA1、SPTB及EPB42,这些基因分别编码锚蛋白、带3蛋白、α收缩蛋白、β收缩蛋白及4.2蛋白。本研究对10例诊断为HS患儿的临床资料进行总结,并且采用高通量测序技术进一步行基因检测,旨在辅助诊断,提高临床中不明原因溶血性贫血的病因诊断,减少误诊与漏诊;同时,丰富HS致病基因库数据,为优生优育提供遗传学参考。现将研究结果报道如下。
选择2017年1月1日至2019年7月31日,于昆明市儿童医院门诊及住院诊断为HS的10例患儿为研究对象。其中,女性患者为3例,男性为7例,平均年龄为4.5岁。本研究纳入标准:符合HS的诊断标准[2],并且接受高通量测序技术检测。排除标准:不愿参与本研究者。本研究遵循的程序符合2013年修订版《世界医学协会赫尔辛基宣言》的要求。
本研究HS的诊断参照《儿科学》第9版相关标准进行[2]。HS临床表现以贫血、黄疸、脾大为主,并且外周血球形红细胞比例增多,红细胞渗透脆性增加。
每例患儿均完善血常规检查,外周血红细胞形态,红细胞渗透脆性试验等检查。并且进行地中海贫血基因,铁蛋白水平,红细胞葡萄糖-6-磷酸脱氢酶(glucose-6-phosphate-dehydrogenase,G-6-PD)水平检测,以及胸部X线摄片。
采集患儿静脉全血标本送至北京迈基诺基因科技股份有限公司(北京迈基诺医学检验所)进行高通量测序。采用高通量测序技术,选择血液系统红系相关基因检测组合,对患者进行相关基因突变筛查。本研究中致病基因位点致病性判断方法,严格按照美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南[3]进行。采用Sanger法测序对部分患儿父母相关基因突变进行验证。将患儿的基因突变筛查结果,录入人类基因突变数据库(Human Gene Mutation Database,HGMD)专业版(http://www.hgmd.cf.ac.uk/ac/index.php)进行检索,判断是否为新发基因突变。
给予患儿对症治疗。在血红蛋白(hemoglobin,Hb)值<90 g/L时,给予患儿输注同型悬浮红细胞或者洗涤红细胞。由于4例患儿中度脾大,需要多次输血,并且年龄>3岁,遂对患儿进行部分脾动脉栓塞术。6例患儿未接受部分脾动脉栓塞术治疗。治疗后对患儿进行血常规检查,并且记录输血频次,以评价疗效。
采用回顾性分析的方法,对HS患儿的发病年龄,病程、临床表现进行分析。并且分析患儿高通量基因测序结果。
10例患儿均以贫血、黄疸、脾大为临床表现。患儿贫血以小细胞性贫血为主,重度贫血为4例,中度贫血为4例,轻度贫血为2例。患儿脾大以轻、中度为主,轻度脾大为3例,中度脾大为6例,无一例为重度脾大。患儿输血次数不一,输血次数最多的是2~3个月输注1次。3例患儿外周血球形红细胞比例增多,其余7例正常,4例红细胞渗透脆性增加,其余6例正常。患儿地中海贫血基因正常。患儿铁蛋白水平和G-6-PD水平均正常。胸部X线摄片未见异常表现。10例患儿中,1例患儿父亲年轻时有贫血、脾大的症状,接受脾切除术后症状获得改善,其余患儿家属均无相关临床症状。
高通量基因测序结果显示,10例患儿中6例存在ANK1、2例(兄弟)存在SPTA1、1例存在SPTB基因突变,1例SPTB基因2~3号外显子疑似重复。其中,ANK1基因c.4585C>T(p.R1529X)、c.3877C>T(p.R1293X)突变在HGMD专业版已有文献报道与球形红细胞增多症(spherocytosis)相关;ANK1基因c.1471C>T(p.Q491X)、c.1817delT(p.L606Cfs*31)、c.4390+5G>T(IVS36dsG-T+5)、c.4189C>T(p.L1397F)突变未见文献报道。根据ACMG指南,ANK1基因c.1471C>T(p.Q491X)(图1A)、考虑c.1817delT(p.L606Cfs*31)突变(图1B)为新发可疑致病突变。SPTA1基因c.4766G>A(p.W1589X)、c.5432G>A(p.R1811Q)突变,SPTB基因2~3号外显子疑似重复未见文献报道。其中,考虑SPTA1基因c.4766G>A(p.W1589X)突变(图1C)为新发可疑致病突变。SPTB基因c.4735C>T(p.R1579X)突变已有文献报道。SPTB基因c.797_798del(p.I266Nfs*28)突变(图1D)未见文献报道,并且考虑为新发可疑致病突变。本研究10例HS患儿检出的基因突变,见表1。
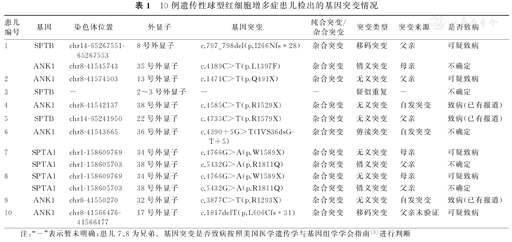
10例遗传性球型红细胞增多症患儿检出的基因突变情况
10例遗传性球型红细胞增多症患儿检出的基因突变情况
| 患儿编号 | 基因 | 染色体位置 | 外显子 | 基因突变 | 纯合突变/杂合突变 | 突变类型 | 突变来源 | 是否致病 |
|---|---|---|---|---|---|---|---|---|
| 1 | SPTB | chr14-65267551-65267553 | 8号外显子 | c.797_798del(p.I266Nfs*28) | 杂合突变 | 移码突变 | 父亲 | 可疑致病 |
| ANK1 | chr8-41545743 | 35号外显子 | c.4189C>T(p.L1397F) | 杂合突变 | 错义突变 | 母亲 | 不确定 | |
| 2 | ANK1 | chr8-41574503 | 13号外显子 | c.1471C>T(p.Q491X) | 杂合突变 | 无义突变 | 父亲 | 可疑致病 |
| 3 | SPTB | - | 2~3号外显子 | - | - | 疑似重复 | - | 不确定 |
| 4 | ANK1 | chr8-41542137 | 38号外显子 | c.4585C>T(p.R1529X) | 杂合突变 | 无义突变 | 自发突变 | 致病(已有报道) |
| 5 | SPTB | chr14-65241950 | 22号外显子 | c.4735C>T(p.R1579X) | 杂合突变 | 无义突变 | 父亲 | 致病(已有报道) |
| 6 | ANK1 | chr8-41543665 | 36号外显子 | c.4390+5G>T(IVS36dsG- T+5) | 杂合突变 | 剪接突变 | 自发突变 | 不确定 |
| 7 | SPTA1 | chr1-158609769 | 34号外显子 | c.4766G>A(p.W1589X) | 杂合突变 | 无义突变 | 母亲 | 可疑致病 |
| SPTA1 | chr1-158605703 | 38号外显子 | c.5432G>A(p.R1811Q) | 杂合突变 | 错义突变 | 父亲 | 不确定 | |
| 8 | SPTA1 | chr1-158609769 | 34号外显子 | c.4766G>A(p.W1589X) | 杂合突变 | 无义突变 | 母亲 | 可疑致病 |
| SPTA1 | chr1-158605703 | 38号外显子 | c.5432G>A(p.R1811Q) | 杂合突变 | 错义突变 | 父亲 | 不确定 | |
| 9 | ANK1 | chr8-41550270 | 32号外显子 | c.3877C>T(p.R1293X) | 杂合突变 | 无义突变 | 自发突变 | 致病(已有报道) |
| 10 | ANK1 | chr8-41566476-41566477 | 17号外显子 | c.1817delT(p.L606Cfs*31) | 杂合突变 | 移码突变 | 父亲未验证 | 可疑致病 |
注:"-"表示暂未明确;患儿7、8为兄弟。基因突变是否致病按照美国医学遗传学与基因组学学会指南[3]进行判断
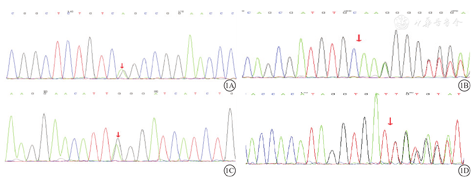
注:箭头示突变位点
本研究4例接受部分脾动脉栓塞术患儿中,2例患儿(患儿7、9)术后,未再接受输血治疗,Hb值维持在约100 g/L;1例(患儿8)术后6个月内,未再接受输血治疗,6个月后每2~3个月接受1次输血治疗;1例(患儿6)术后失访。未接受部分脾动脉栓塞术治疗的6例患儿中,4例患儿(患儿1、3、5、10)未合并感染情况下,Hb值>90 g/L,波动于90~108 g/L;合并感染时,Hb值下降至<90 g/L,感染控制后,Hb值可自行恢复至感染发生前的水平,未给予其他特殊治疗;1例患儿(患儿4)给予补充铁剂后,Hb值升至约100 g/L,未再予输血治疗;1例患儿(患儿2)给予口服铁剂、叶酸无效,仍定期每2个月输血1次。
典型的HS根据溶血表现、外周血检出球形红细胞,以及家族史,不难诊断。但是若患者临床表现不典型,外周血无异常形态红细胞,无家族史的患者容易误诊、漏诊。HS患者外周血球形红细胞比例差别较大,最少为1%~2%,最多可达60%~70%[4]。本研究对10例患儿的临床资料进行分析,发现全部患儿均存在不同程度的贫血、黄疸、脾大的表现,其中3例外周血球形红细胞比例增多,其余7例正常,4例红细胞渗透脆性增加,其余6例正常,仅1例有家族史。采用高通量测序技术发现,4种HS新发可疑致病基因突变,包括ANK1基因c.1471C>T(p.Q491X)、c.1817delT(p.L606Cfs*31)突变,SPTA1基因c.4766G>A(p.W1589X)突变,SPTB基因c.797_798del(p.I266Nfs*28)突变。因此,对于反复贫血、黄疸、有输血病史、脾大的患者,相关药物治疗无效,并且排除地中海贫血,骨髓检查结果为增生性贫血的患者,可进行红细胞相关基因突变检查,以辅助诊断。
HS为最常见的遗传性红细胞膜骨架异常,使红细胞形态发生改变、变形性下降,在单核-巨噬细胞系统内被阻留破坏,临床表现为溶血、黄疸、脾大。HS最早在1871年被报道,多见于北美与北欧,在黑色人种中发病率低,在非洲、日本、巴西、北印度等也有文献报道,在我国的发病率为1∶100 000[5],我国各地均有文献报道。HS最有效的治疗是脾切除术,但是一般要求患儿年龄>6岁。HS患儿经脾切除术后,免疫功能显著下降,易发生反复感染。因此,HS患儿在接受脾切除术前,应完善多种疫苗的接种,术后长期口服抗菌药物预防感染[6]。目前,昆明市儿童医院采取部分脾动脉栓塞术,通过肿大的脾部分缺血性坏死,从而使脾缩小,改善HS患儿的临床溶血症状。该方法可使部分患儿脱离反复输血,避免输血不良反应的发生风险,并且减轻反复输血造成的经济负担,提高患儿的生活质量。本研究中,2例患儿接受部分脾动脉栓塞术后,未再接受输血治疗,Hb值可维持在约100 g/L。但是部分脾动脉栓塞术的缺点与部分脾切除术一样具有复发风险。因此,临床医师要结合HS患儿具体情况,权衡利弊采取有效的治疗手段。
HS的发生与红细胞无法维持正常的双凹形,导致过早被机体清除相关。目前已知的抗红细胞弹性变形的结构是由蛋白质网络、脂质双层及跨膜蛋白质相互交联形成[7]。而锚蛋白与带3蛋白、收缩蛋白与4.2蛋白连接稳定细胞膜[8]。ANK1基因位于8号染色体短臂11.1,ANK1突变为HS最常见的原因,其次为SLC4A1与SPTB基因突变,主要基因突变ANK1、SLC4A1、SPTB基因突变可导致对应氨基酸改变,引起蛋白缺乏[9]。ANK1基因突变同时存在常染色体显性遗传与常染色体隐性遗传2种遗传模式。在HS中,绝大多数基因突变是新发突变[10]。目前在HGMD中已报道59种不同ANK1基因突变,包括缺失、移码、无义或者错义突变[11]。HS临床严重程度不一,可无症状,亦可表现为危及生命的贫血[12]。伴SPTB基因突变HS患儿发生再生障碍性危象的风险高于伴ANK1基因突变者[13]。
多数HS患儿在儿童或者青少年时期就可以获得诊断,但是对于无症状或者轻微症状的患者,通常是在患儿发生再生障碍性危象或者体检才发现。对于临床症状不典型或是有临床症状,但是常规实验室检查不能诊断时,或者没有家族史的患儿,容易出现漏诊或者误诊[14]。因此,需要进行相关基因检测,二代基因测序(next-generation sequencing,NGS)已广泛应用于诊断遗传性红细胞膜病[15]。本研究通过高通量测序发现4个暂无文献报道的ANK1、SPTA、SPTB基因新发突变。丰富了HS基因突变数据库,为进一步辅助HS诊断及研究HS的病因提供参考。
所有作者均声明不存在利益冲突

























