
Maffucci综合征是一种以全身多发性内生软骨瘤伴发血管瘤为特征的罕见疾病。本文就1例IDH1突变的24岁女性Maffucci综合征患者病例报告进行临床分析,并复习国内外文献,提高对该病的认识。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Maffucci综合征(Maffucci syndrome)是一种罕见的非遗传性中胚层发育不良疾病,表现为多发内生软骨瘤合并真皮、皮下组织或内脏的多发血管瘤。本文就1例IDH1突变的Maffucci综合征病例报告进行临床分析,并复习国内外文献,加强对该病的认识,为Maffucci综合征的诊治提供更多信息。
病例资料:患者女性,24岁,因"发现手掌、手指、右上肢形态异常8年"于2018年8月就诊于本院门诊。患者2010年出现双侧指关节处多发形态异常,表现为不规则隆起,约黄豆大小,质硬,局部无压痛,无红肿热痛,无破溃,关节活动受限;右侧肱骨上端亦出现不规则隆起,鸡蛋大小,性质同指关节处的多发隆起,患者未予重视,未规范就诊,上述隆起缓慢增大,于2016年右手掌大鱼际处出现不规则隆起,质韧,豌豆大小,偶有压痛,无破溃,可见血管纹理,后逐渐增大,曾到当地医院就诊,查X线提示代谢性骨病的可能性大。患者为求进一步诊治,来我院就诊,门诊以"骨骼异常查因:代谢性病?"收入我科。患者自起病以来,精神、睡眠、食纳可,大小便正常,体重无明显改变。既往史、个人史、家族史、婚育史无特殊。
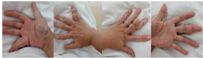
体格检查:身高:150 cm,体重:44 kg,体重指数17.4 kg/m2,脊柱无畸形,棘突无压痛,无叩痛,四肢活动正常。右肩关节、肱骨及双手指关节附近可见不规则隆起,质地硬,无压痛。右手大鱼际处可见肿胀,呈蓝色,质地软,无搏动感,无震颤,皮温正常,压痛(+),余无明显畸形(图1)。
实验室及辅助检查:血管炎3项、血尿轻链、血钙、磷、酸性磷酸酶、碱性磷酸酶、β胶原降解产物和甲状旁腺激素、降钙素、25-羟基维生素D水平均正常。
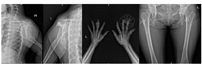
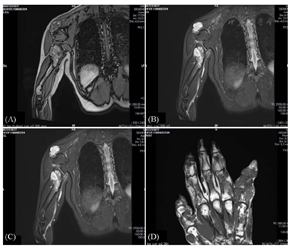
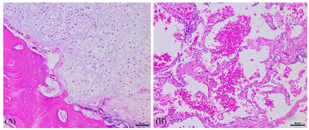
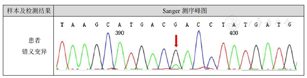
X线检查:双侧肩胛骨、双侧肱骨近段(图2)、右侧第2、5、6前肋、双侧股骨上段及左侧股骨下端(图2)、左胫骨、右侧尺桡骨远段、右手多发掌指骨、左侧第3、4掌指骨见多发骨密度减低,部分呈膨胀性骨质破坏。左侧第3指骨、右侧掌骨周围软组织内见斑点状钙化灶,双侧手指畸形改变(图2),右肱骨缩短畸形,左侧胫骨局部骨质扭曲。右上肢及右手磁共振平扫增强:右侧肩胛骨、右侧肱骨(图3A及图3B)、右侧桡骨中远段、右侧尺骨内侧远端(图3C)右手多发掌指骨、部分腕骨内可见弥漫多发不规则团状、结节状长T1长T2信号灶,部分呈膨胀性骨质破坏,部分掌指及指间关节部分畸形,邻近软组织肿胀、受累,增强后可见明显强化(图3D)。右手大鱼际处超声:皮下软组织层可探及多个低回声结节,形态欠规则,边界欠清,内光点粗,分布欠均匀,结节内可见大小不等的强光团伴声影。与患者及家属协商后在全麻下行右手软组织肿块活检术及右小指肿瘤活检术,病检结果为(右小指肿瘤)骨软骨瘤(图4A)及(右手肿块)血管瘤(图4B),Sanger测序法检测切除的软骨及血管标本中的异柠檬酸脱氢酶(IDH)基因是否有突变,结果发现IDH1 R132C突变(图5)。
根据临床表现、体征、实验室检查、病理检查和基因检测结果,该患者的诊断是:Maffucci综合征。
Maffucci综合征(Maffucci syndrome)是一种以全身多发性内生软骨瘤伴发血管瘤为特征的罕见疾病。Angelo Maffucci在1881年首次描述了一种特征为青少年时期发病的多发性血管瘤和内生软骨瘤的非遗传疾病。此后,该种疾病被称作"Maffucci综合征"。其诊断要点主要涉及全身多发性内生软骨瘤伴发血管瘤,偶尔也可伴发淋巴管瘤。Maffucci综合征的血管和骨骼病变通常是不对称的,50%的患者为单侧病变[1]。该病不影响智力。迄今为止的文献表明,这种综合征可发生于所有种族,发病率没有性别差异。就医时的年龄从6~58岁不等,但这种疾病通常在青春期前发病,后逐渐进展。
内生软骨瘤是一种可发生在任何部位的良性髓内软骨瘤,但最常见于指骨和长骨生长板附近,有时也累及胫骨、腓骨、股骨、肋骨及颅骨等,可导致骨骼畸形、肢长不一致以及病理性骨折。本例患者发病部位在双侧肩胛骨、双侧股骨近段、右侧第2、5、6前肋、左侧胫骨、右侧尺桡骨远段、右手多发掌指骨、左侧第3、4掌指骨,属于该病常见的发病部位。随疾病进展,大约30%~40%的内生软骨瘤可转化为软骨肉瘤[2]。
血管瘤可见于多个部位,包括皮下、黏膜表面、内脏器官、呼吸道、胃肠道及主动脉等,以肢端浅表部位多见,通常表现为海绵状血管瘤、静脉扩张、淋巴管瘤。本例患者血管瘤发病部位在双手指,也属于该病常见的发病部位。血管瘤多表现为受压压缩的、无痛性蓝色结节。发生于消化道的血管瘤可导致出血所致的难治性缺铁性贫血,而上消化道或喉部的病变破裂可引起呼吸窘迫和吞咽困难等症状[3,4]。既往国内外病例报道脾脏受累的发生率约在20%左右,表明脾脏很可能是Maffucci综合征内脏血管病变的好发部位[5]。此外,至今为止没有证据提示血管瘤会发生内脏转移,也没有患者死于转移性血管内皮瘤的报道[6]。
除此之外,Maffucci综合征患者中其他肿瘤的发病率也有所增加,包括发生于胰腺、垂体、肾上腺、甲状腺和甲状旁腺的纤维瘤、腺瘤和腺癌,以及卵巢畸胎瘤、胶质瘤、胶质母细胞瘤、急性髓性白血病和肝内胆管细胞癌等[7,8,9,10,11,12,13]。
本病的诊断需结合临床表现,主要依据活检病理组织,发现同时存在软骨瘤及血管瘤,同时病变部位X线检查,对诊断Maffucci综合征至关重要。X线可见边界清晰的可透射线的骨骼损伤及受累骨骼皮质变薄和骨内膜扇贝样变[14]。周围软组织可有多处小钙化,提示有静脉石形成。当怀疑是恶性变时,应进行活检以明确诊断。而磁共振成像(MRI)扫描有助于减少因肿瘤异质性引起的采样误差[15]。
该病的病因目前仍在研究中。目前已有文献报道IDH基因的体细胞突变与Maffucci综合征相关,Pansuriya等[16]发现77%的Maffucci综合征患者携带IDH1和(或)IDH2突变。人类IDH有三种类型,分别为IDH1、IDH2和IDH3。IDH1定位与细胞质和过氧化物酶体中,IDH2和IDH3定位于线粒体中。其中IDH1基因位于染色体2q34,包含10个外显子,编码414个氨基酸。IDH1基因编码胞浆内烟酰胺腺嘌呤二核苷酸磷酸依赖的异柠檬酸脱氢酶,后者能够将异柠檬酸进行催化生成α-酮戊二酸。作为一种编码代谢关键酶的基因,IDH1基因突变可以将α-酮戊二酸转变成2-羟基戊二酸(2-hydroxyglutarate,2HG),从而抑制TET2酶,导致甲基化增加,促进细胞增殖和促进肿瘤发生[17]。
已有研究表明,在87%的内生软骨瘤、良性软骨瘤和70%的梭形细胞血管瘤(良性血管病变)中也发现了IDH1和IDH2突变[16]。IDH突变可导致小鼠体内的软骨细胞的持续存在,形成内生软骨瘤[18]。这种突变可能是由于体细胞嵌合形成[19]。IDH1或IDH2的杂合子体细胞突变在其他恶性肿瘤的发展中也同样不可或缺[7]。绝大多数Ⅱ级和Ⅲ级(WHO分级)的胶质瘤和继发性胶质母细胞瘤中都发现了编码IDH1和IDH2基因的体细胞突变[11]。此外大约10%的成人新发性正常核型急性髓系白血病(acute myeloid leukemia,AML)中,也发现了IDH1或IDH2突变[20]。这些研究表明,Maffucci综合征易伴发肿瘤的风险增加是由于共同的遗传背景——起始于胚胎发育的IDH1或IDH2镶嵌突变引起的。
通过Sanger测序技术检测,以及一代测序验证,本例患者组织样本存在IDH1基因1个错义突变。该突变导致IDH1基因第132位密码子编码的氨基酸由精氨酸变为半胱氨酸。该突变为罕见突变,没有被gnomAD数据库东亚人群、EXAC数据库或千人基因组数据库收录;SIFT和MutationTaster等多个软件预测该突变对基因或基因产物产生有害影响。已有多篇文献报道在Maffucci综合征患者体细胞中检测到该突变,并提出该突变为IDH1基因突变热点[21,22];该突变已被COSMIC(Catalogue of Somatic Mutations in Cancer)数据库收录。
目前,Maffucci综合征尚无有效的药物治疗。为缓解疼痛及早期诊治恶变,手术仍是唯一的选择。而手术干预只适用于正畸整形、骨折或相关并发症,如血管瘤病变的恶性转化和结缔组织变性。骨性病变可行手术切除或刮除肿瘤并填充,血管病变可行手术、硬化或放射治疗[23]。因为该病的恶变可能性大,那么Maffucci综合征的及时诊治和终身监测就十分重要。为了及早诊断软骨肉瘤,Vedegaal等[9]建议对多发内生软骨瘤的患者的每个内生软骨瘤都应行X线检查,以便将来进行比对。Schwartz等[23]对44例内生软骨瘤患者进行了研究,7例诊断为Maffucci综合征,37例诊断为Ollier综合征。在他们的研究中,没有一个患者死于骨骼肉瘤,但是在该研究中,5例非骨骼恶性病变的患者中有4例死亡。因此,对于非骨骼肿瘤的检测也十分重要。一些作者建议在出现神经或腹部症状时进行大脑或腹部CT检查[9]。但是,当出现临床表现时,恶性病变的治疗可能已经为时已晚[7]。
综上所述,Maffucci综合征是一种以多发性内生软骨瘤和血管瘤为特征的罕见病,需与之鉴别的是仅表现为多发内生软骨瘤的Ollier综合征,同时还要注意合并亚临床(即组织学上存在但临床上不明显)、轻微的血管瘤合并多发内生性软骨瘤患者。除了及时诊断,还应注意对病变处及症状的监测,以早期发现恶性肿瘤,及早治疗,改善患者预后。
所有作者均声明不存在利益冲突

























