分析先天性纯红细胞再生障碍性贫血(PRCA),即Diamond-Blackfan贫血(DBA)患儿的临床表现、实验室检查、基因检测结果、疗效及生长发育评估结果,为其诊断与治疗提供更多的临床思路。
选择2011年4月至2019年2月于郑州大学第一附属医院诊治的25例DBA患儿为研究对象。其中男性患儿为13例,女性为12例,中位发病年龄为3个月。按照治疗方法不同,将其分为激素治疗组(使用泼尼松治疗,n=18)和非激素治疗组(使用对症支持治疗,n=7)。采用回顾性研究方法,收集患儿一般临床资料、实验室检查结果、疗效、生长发育评估结果等。2组间缓解率等比较,采用Fisher确切概率法。不同疗程、年龄患儿身高、体重百分位比较,采用Kruskal-Wallis秩和检验。本研究遵循的程序符合2013年修订的《世界医学协会赫尔辛基宣言》要求。
①本研究25例DBA患儿的中位发病年龄为3个月,男、女性别构成比为1.08∶1。伴先天性疾病患儿为5例(20%),其中4例患儿合并≥2种先天畸形。②血常规结果显示,患儿血红蛋白(Hb)值均降低,64%(16/25)患儿网织红细胞计数减少。骨髓细胞学检查显示,患儿均有核细胞增生活跃,红系增生低下。12例行Hb电泳患儿中,胎儿血红蛋白(HbF)增高患儿为2例。13例行基因检测患儿中,4例患儿DBA相关基因突变,包括RPS19基因突变者3例和RPL5基因突变者1例。③激素治疗组和非激素治疗组患儿缓解率分别为88.9%(16/18)和42.9%(3/7),2组比较,差异有统计学意义(P=0.032)。④ 20例DBA患儿生长发育评估结果显示,短疗程激素治疗者(激素治疗时间<6个月,n=7)、长疗程激素治疗者(激素治疗时间≥6个月,n=8)与未使用激素治疗者(n=5)在治疗远期体重、身高百分位数的中位数分别比较,差异均无统计学意义(χ2=2.456,P=0.293; χ2=0.460,P=0.795);小年龄激素治疗者(接受激素治疗时患儿年龄<12个月,n=8),大年龄激素治疗者(接受激素治疗时患儿年龄≥12个月,n=7)与未使用激素治疗者(n=5)治疗远期体重、身高百分位数的中位数分别比较,差异亦无统计学意义(χ2=1.390,P=0.499; χ2=0.624,P=0.732)。
DBA通常于出生早期发病,其诊断需依靠血常规及骨髓细胞学检查。DBA患儿中RPS19及RPL5基因突变较常见,相关基因检测有利于该病的早期诊断和激素治疗。DBA患儿身高、体重的生长发育情况与是否使用激素及激素使用疗程,以及接受激素治疗时患儿年龄无显著关系。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
纯红细胞再生障碍性贫血(pure red cell aplasia,PRCA)是由于骨髓中红细胞系增殖分化障碍,幼红细胞数量显著减少或缺如所致的一组骨髓衰竭性疾病,该病不影响患者粒系及巨核系细胞增殖。根据病因,PRCA可分为先天性和获得性PRCA,后者又可分特发性与继发性PRCA。先天性PRCA亦被称为Diamond-Blackfan贫血(Diamond-Blackfan anemia, DBA),该病患儿发病年龄较小,生后即可出现贫血表现,>90% DBA患儿在出生后1年内发病,可伴有先天畸形,并且具有肿瘤易感性[1]。DBA的确切发病原因目前仍未明确,可能与核糖体蛋白基因突变相关。糖皮质激素为DBA首选治疗药物,但部分患儿对糖皮质激素不敏感或需终身用药。为提高临床医师对DBA的认识及临床诊治水平,现对2011年4月至2019年2月于郑州大学第一附属医院诊治的25例DBA患儿的病例资料进行回顾性分析。现将研究结果报道如下。
选择2011年4月至2019年2月于郑州大学第一附属医院诊治的25例DBA患儿为研究对象。其中,男性患儿为13例,女性为12例,中位发病年龄为3个月。按照治疗方法不同,将研究对象分为激素治疗组(使用泼尼松治疗,n=18)和非激素治疗组(使用对症支持治疗,n=7)。2组患儿年龄、性别构成比等基本临床资料比较,差异均无统计学意义(P>0.05)。本研究遵循的程序符合2013年修订的《世界医学协会赫尔辛基宣言》要求。
研究对象纳入标准:①符合DBA诊断标准[2];②具有完整的临床资料及随访资料。排除标准:①感染、肿瘤、药物或毒物等所致的急性造血功能停滞;②恶性血液性疾病、结缔组织病、慢性溶血性疾病、自身免疫性疾病等可能导致单纯红细胞减少的疾病。
DBA诊断及分型标准参照2008年第6届Daniella Maria Arturi国际年会制定的诊断标准[2]。DBA的诊断指标,见表1。

Diamond-Blackfan贫血的诊断指标
Diamond-Blackfan贫血的诊断指标
| 项目 | 临床表现 |
|---|---|
| 主要诊断标准 | ①发病年龄<1岁 |
| ②大细胞性贫血,粒细胞及血小板无明显减少 | |
| ③网织红细胞明显减少 | |
| ④骨髓增生正常,伴红系前体细胞减少或缺乏 | |
| 主要支持标准 | ①DBA致病基因突变 |
| ②阳性家族史 | |
| 次要支持标准 | ①红细胞腺苷脱氨酶活性升高 |
| ②躯体畸形 | |
| ③HbF值升高 | |
| ④无其他先天性骨髓衰竭性疾病证据 |
注:经典型DBA的诊断需满足全部4条主要诊断标准。非经典DBA的诊断需满足任意一条主要支持标准,或者满足任意一条主要诊断标准+DBA致病基因突变。拟诊DBA的诊断需满足3条主要诊断标准及阳性家族史;同时满足2条主要诊断标准+3条次要支持标准;满足阳性家族史+3条次要支持标准。DBA为Diamond-Blackfan贫血,HbF为胎儿血红蛋白
激素治疗组DBA患儿接受口服泼尼松片治疗,剂量为2.0 mg/(kg·d),分为3次服用;足量治疗至少1个月,并且患儿血红蛋白(hemoglobin,Hb)值≥100 g/L时,开始逐渐减量至1.0 mg/(kg·d);维持治疗2~3个月,并使Hb值为90~100 g/L,然后,根据患儿Hb值缓慢减量至停药。非激素治疗组患儿给予对症支持处理,若Hb值≤60 g/L则给予输血治疗。
本研究疗效评价标准[2]如下。完全缓解(complete remission,CR)定义为治疗1个月后,Hb值≥90 g/L且不依赖输血。部分缓解(partial remission,PR)定义为治疗期间Hb值为80~90 g/L。治疗无效定义为开始治疗1个月内,Hb值仍呈进行性下降并依赖输血,或者激素减量后Hb值<80 g/L。缓解后复发定义为激素治疗第1个月达CR,停药后Hb值≤80 g/L,并且排除其他疾病可能。缓解率计算公式为:缓解率(%)=(CR+PR)患儿例数/患儿总例数×100%
收集患儿一般临床资料,包括患儿诊断年龄、性别、首发症状、先天畸形、阳性家族史等。记录实验室检查结果,包括血常规、初诊时Hb值、骨髓细胞学检查、Hb电泳结果、DBA相关基因(RPS19、RPS24、RPS17、RPL35A、RPL5、RPL11、RPS7、RPS10、RPS26、RPL26等基因)检测结果等。
记录25例患儿的出生体重,以及其治疗远期(激素治疗组停药后6个月和非激素治疗组对症支持治疗6个月后)的身高及体重。根据中国0~18岁儿童身高、体重的百分位数标准值对照表[3],获得患儿不同观察时间点身高、体重百分位数。生长发育评估均需排除存在以下情况的患儿:①父母或直系亲属中存在男性身高<160 cm,女性身高<150 cm,无法排除矮小症可能(出生体重、身高<同龄儿75%百分位数;②合并其他影响生长发育的疾病;③除激素外,同时应用其他可能影响生长发育的药物。
通过门诊及电话对患儿进行随访。随访截至时间为2019年12月1日。随访频率:初诊治疗后1个月随访1次,对激素治疗组患者,在维持治疗期间每月随访1次,停药后每6个月随访1次。随访内容包括患儿外周血血常规、身高及体重。全部患儿中位随访时间为43个月(9~81个月)。
本研究所得数据采用SPSS 21.0软件进行统计学处理。CR率、PR率、缓解率等计数资料采用百分比(%)表示,组间比较采取Fisher确切概率法。患儿身高、体重百分位数的中位数采用M(P25~P75)表示,组间比较采用Kruskal-Wallis秩和检验。本研究所有统计学检验采用双侧检验,以P<0.05表示差异有统计学意义。
本研究25例DBA患儿的中位发病年龄为3个月,发病年龄≤3个月患儿为13例(52%),≤6个月患儿为17例(68%),≤1岁患儿为24例(96%);男、女性别构成比为1.08∶1。患儿首发症状均为贫血,无出血、反复感染及淋巴结大表现,出现轻度肝、脾大患儿为6例(24%)。伴先天畸形患儿为5例(20%,4例患儿合并2种及以上先天畸形)。其中,伴先天性心脏病患儿为4例,手指畸形为2例,右侧睾丸未降为1例,先天性腭裂合并先天性脊柱裂为1例。25例DBA患儿中,存在贫血家族史患儿为3例(12%),其中1例患儿胞兄因PRCA于生后3个月夭折,另一胞兄为死胎。25例患儿均无前驱感染史、急性溶血及自身免疫性疾病等。
本研究25例患儿血常规结果示,Hb值均呈不同程度下降,初诊Hb值为16.2~84.0 g/L;网织红细胞计数减少患儿为16例(64%),网织红细胞百分比为0.1%~1.2%;大细胞性贫血患儿为5例(20%),其余患儿均为正细胞正色素性贫血。骨髓细胞学检查显示,患儿均存在有核细胞增生活跃,红系增生低下,红系比例为0~4.8%,粒系、巨核系正常。25例患儿中,12例患儿行Hb电泳,结果显示,胎儿Hb(fetal Hb,HbF)增高患儿为2例,其余13例患儿未行该检查。13例行基因检测患儿中,检出DBA相关基因突变患儿为4例(表2),其中3例患儿为RPS19基因突变,1例患儿为RPL5基因突变,其余12例患儿未行该检查。
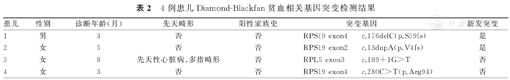
4例患儿Diamond-Blackfan贫血相关基因突变检测结果
4例患儿Diamond-Blackfan贫血相关基因突变检测结果
| 患儿 | 性别 | 诊断年龄(月) | 先天畸形 | 阳性家族史 | 突变基因 | 新发突变 | |
|---|---|---|---|---|---|---|---|
| 1 | 男 | 3 | 否 | 否 | RPS19 exon4 | c.176delC(p.S59fs) | 是 |
| 2 | 女 | 5 | 否 | 否 | RPS19 exon2 | c.13dupA(p.V4fs) | 是 |
| 3 | 女 | 8 | 先天性心脏病、多指畸形 | 否 | RPL5 exon3 | c.189+1G>T | 否 |
| 4 | 女 | 3 | 否 | 否 | RPS19 exon4 | c.280C>T(p.Arg94) | 否 |
激素治疗组CR率为72.2%(13/18),PR率为16.7%(3/18),治疗无效率11.1%(2/18)。非激素治疗组CR率为42.9%(3/7),治疗无效率57.1%(4/7)。激素治疗组和非激素治疗组缓解率分别为88.9%(16/18)和42.9%(3/7),2组比较,差异有统计学意义(P=0.032)。
本研究25例患儿中,排除疑似矮小症患儿2例,以及合并复杂先天性心脏病患儿3例后,进行生长发育评估患儿共计20例。20例不同化疗疗程和治疗方案DBA患儿生长发育评估结果显示,短疗程激素治疗者(激素治疗时间<6个月,n=7)、长疗程激素治疗者(激素治疗时间≥6个月,n=8)与未使用激素治疗者(n=5)在治疗远期的体重、身高百分位数的中位数分别比较,差异均无统计学意义(χ2=2.456,P=0.293;χ2=0.460,P=0.795),见表3;小年龄激素治疗者(接受激素治疗时患儿年龄<12个月,n=8)、大年龄激素治疗者(接受激素治疗时患儿年龄≥12个月,n=7)与未使用激素治疗者(n=5)在治疗远期的体重、身高百分位数的中位数分别比较,差异亦无统计学意义(χ2=1.390,P=0.499; χ2=0.624,P=0.732),见表4。
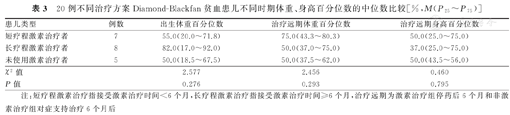
20例不同治疗方案Diamond-Blackfan贫血患儿不同时期体重、身高百分位数的中位数比较[%,M(P25~P75)]
20例不同治疗方案Diamond-Blackfan贫血患儿不同时期体重、身高百分位数的中位数比较[%,M(P25~P75)]
| 患儿类型 | 例数 | 出生体重百分位数 | 治疗远期体重百分位数 | 治疗远期身高百分位数 |
|---|---|---|---|---|
| 短疗程激素治疗者 | 7 | 55.0(20.0~71.8) | 75.0(43.3~80.3) | 50.0(25.0~75.0) |
| 长疗程激素治疗者 | 8 | 82.0(17.0~92.0) | 50.0(37.0~75.0) | 37.0(25.0~75.0) |
| 未使用激素治疗者 | 5 | 50.0(18.5~67.5) | 50.0(37.5~62.0) | 50.0(43.5~56.0) |
| χ2值 | 2.577 | 2.456 | 0.460 | |
| P值 | 0.276 | 0.293 | 0.795 |
注:短疗程激素治疗指接受激素治疗时间<6个月,长疗程激素治疗指接受激素治疗时间≥6个月,治疗远期为激素治疗组停药后6个月和非激素治疗组对症支持治疗6个月后
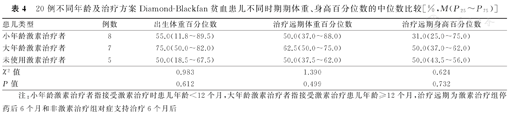
20例不同年龄及治疗方案Diamond-Blackfan贫血患儿不同时期期体重、身高百分位数的中位数比较[%,M(P25~P75)]
20例不同年龄及治疗方案Diamond-Blackfan贫血患儿不同时期期体重、身高百分位数的中位数比较[%,M(P25~P75)]
| 患儿类型 | 例数 | 出生体重百分位数 | 治疗远期体重百分位数 | 治疗远期身高百分位数 |
|---|---|---|---|---|
| 小年龄激素治疗者 | 8 | 55.0(11.8~89.5) | 50.0(37.0~88.0) | 31.0(25.0~75.0) |
| 大年龄激素治疗者 | 7 | 75.0(50.0~82.0) | 62.5(50.0~75.0) | 50.0(37.0~62.0) |
| 未使用激素治疗者 | 5 | 50.0(18.5~67.5) | 50.0(37.5~62.0) | 50.0(43.5~56.0) |
| χ2值 | 0.983 | 1.390 | 0.624 | |
| P值 | 0.612 | 0.499 | 0.732 |
注:小年龄激素治疗者指接受激素治疗时患儿年龄<12个月,大年龄激素治疗者指接受激素治疗患儿年龄≥12个月,治疗远期为激素治疗组停药后6个月和非激素治疗组对症支持治疗6个月后
DBA是一种罕见的先天性骨髓衰竭性疾病,其主要特点是骨髓红系造血衰竭伴先天性畸形,以及肿瘤发生风险增加[2,4]。DBA的确切发病率目前仍未明确,国外研究结果显示,欧美国家DBA发病率为(5~7)×10-6,日本为12×10-6,大部分为散发病例[5,6];国内目前尚无DBA的流行病学调查资料。有研究表明,约90%的DBA患儿发病年龄<1岁,10%~25%患儿具有阳性家族史,临床以贫血(大细胞性贫血)为主要表现[1]。本研究结果显示,25例DBA患儿的中位发病年龄为3个月,发病年龄≤3个月患儿为13例(52%),≤6个月患儿为17例(68%),≤1岁患儿为24例(96%);男、女性别构成比为1.08∶1;25例患儿均以贫血为主要表现,无明显前驱感染史,无基础疾病。以上结果与国外研究结果[2]相符。
国外研究结果表明,DBA患儿常伴各种先天性发育异常,包括颅面器官畸形、指/趾畸形、泌尿生殖系统畸形、心血管系统畸形、肌肉骨骼系统畸形、运动神经系统畸形等[7]。本研究5例DBA患儿伴发先天畸形,其中3例患儿合并≥2种先天畸形;伴先天性心脏病患儿为4例,并指畸形为2例,右侧睾丸未降为1例,先天性腭裂合并先天性脊柱裂为1例。该结果与DBA伴发先天发育异常表现相符[7]。
DBA患儿实验室检查结果常显示,网织红细胞计数减少或缺如,骨髓增生活跃,粒系、巨核系增生正常,红系增生明显缺乏[1]。本研究结果示,25例患儿初诊Hb值为16.2~84.0 g/L,网织红细胞百分比为0.1%~1.2%;大细胞性贫血患儿为5例,其余患儿均为正细胞正色素性贫血。骨髓细胞学检查显示,患儿有核细胞增生活跃,红系增生低下,红系比例为0~4.8%,粒系、巨核系正常。12例行Hb电泳患儿中,HbF增高患儿为2例。
目前,DBA被认为是一种核糖体合成障碍或功能缺陷型疾病[8,9],病因与发病机制较复杂,可能与遗传、免疫因素有关,多呈常染色体显性或隐性遗传。核糖体是细胞内催化蛋白合成的细胞器,由1个小亚单位(小亚基,40S)和1个大亚单位(大亚基,60S)组成,这些亚单位又由80种结构不同的蛋白质组成[10]。编码核糖体小亚基蛋白的RPS19基因首先被证实与DBA发病相关。随后,研究者陆续发现与DBA相关的其他核糖体蛋白基因,包括RPSl7、RPL5、RPS24等[8,11,12,13]。有研究结果显示,RPS19基因突变,导致核糖体蛋白单倍型不足、核糖体功能异常,而导致P53蛋白异常增多[14],过多的P53蛋白可通过增加肿瘤坏死因子(tumor necrosis factor,TNF)-α,激活p38丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路,降低珠蛋白转录因子(globin transcription factor)-1表达等多种方式,引起早期红系祖细胞凋亡。本研究结果亦显示,13例行基因检测患儿中,检出DBA相关基因突变患儿为4例(表2),其中3例患儿为RPS19基因突变,1例患儿为RPL5基因突变。但是,目前DBA的诊断标准仅基于临床特征和血液学检查结果,而其他骨髓衰竭性疾病及病毒感染相关性贫血亦可出现类似表现,因此DBA特异性生化检测手段的缺乏,为明确DBA诊断造成一定困难。此外,伴相关基因阳性的DBA患儿与未检测到典型基因突变患儿的发病机制是否相同,仍有待进一步研究。
DBA的治疗应以维持患儿生长发育所需的Hb值(80~100 g/L)为目的,糖皮质激素是DBA患儿的首选药物[7,15]。本组研究中,激素治疗组和非激素治疗组缓解率比较(88.9%比42.9%),差异无统计学意义(P=0.032)。这提示,DBA仍以糖皮质激素治疗为首选治疗方法。
此外,输血也是DBA患儿的重要治疗方法之一,尤其是小婴儿及对激素无效的DBA患儿。一般应使患儿的Hb值≥80 g/L,以保证患儿的生长发育及重要脏器功能。对于发生输血依赖患儿,应在多次输血后监测有无体内铁超载,及时应用铁螯合剂治疗可预防输血性铁过载发生[16]。
有研究表明,在激素治疗期间DBA患儿会出现生长发育迟缓,可能抑制患儿免疫功能而出现反复感染,因此对于1岁以内的DBA患儿是否应用激素,目前仍有争议[17,18]。本研究对DBA患儿激素治疗后的生长发育情况进行统计分析,结果显示,短疗程激素治疗者、长疗程激素治疗者与未使用激素治疗者在治疗远期的体重、身高百分位数的中位数分别比较,总体差异无统计学意义(χ2=2.456,P=0.293;χ2=0.460,P=0.795)(表3);小年龄激素治疗者、大年龄激素治疗者与未使用激素治疗者在治疗远期的体重、身高百分位数的中位数分别比较,总体差异亦无统计学意义(χ2=1.390,P=0.499;χ2=0.624,P=0.732)(表4)。这提示,激素使用时年龄大小及激素治疗时间长短对治疗远期DBA患儿的身高、体重无显著影响。但是,由于本研究为单中心研究,样本含量小、随访时间较短、患儿家属配合度不高等原因,均可能会影响上述结果。因此,激素给药年龄大小、给药时间长短是否对患儿生长发育有影响,仍需进行大样本量的多中心研究,并且需要记录生长发育情况及相关影响因素,以获得更确切的结论。
目前研究认为,异基因造血干细胞移植(allogeneic hematopoietic stem cell transplantation,allo-HSCT)是DBA唯一治愈方法。美国DBA相关研究结果显示,9岁前接受allo-HSCT的DBA患儿的存活率为(90.0±9.5)%,而对于>9岁上者,存活率为(70.0±11.6)%(P=0.007)[17,19,20]。但是,由于本组DBA患儿年龄较小或有家庭因素等,暂无患儿采取allo-HSCT治疗。
综上所述,DBA是一种罕见的先天性骨髓衰竭性疾病,依靠临床特征、血常规及骨髓细胞学检查,HbF检测有助于诊断。目前基因检测技术可协助诊断,但因技术发展、诊断成本等原因,尚有家属不能接受,对检出率也存在一定的影响。糖皮质激素治疗效果良好,激素无效可间断输血加去铁治疗,有研究表明泼尼松联合环孢素应用可使疾病得到缓解[21]。激素给药年龄大小、给药时间长短对DBA患儿生长发育是否有影响,仍有待扩大样本量进一步研究。
所有作者均声明不存在利益冲突


























