
胆红素是人体一种重要的内源性物质,可以分为结合胆红素和非结合胆红素两大类,全面学习胆红素的产生和运输、胆红素的具体种类及其生物学特性、胆红素的排泄和重吸收等代谢过程,有助于理解临床一系列以黄疸为表现的胆红素异常升高类疾病的发病机制,从而更好地进行相关疾病的诊治工作。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
(1)胆红素的产生:胆红素来源于体内的血红素,其中约70%~80%的血红素产自血循环中衰老的红细胞,经单核-巨噬细胞破坏所降解形成的血红蛋白,少部分来自骨髓幼稚红细胞的血红蛋白、肝内含血红素的蛋白质(如细胞色素P450、细胞色素C、过氧化氢酶等)及肌红蛋白。此时的胆红素称为游离胆红素,又称为未结合胆红素(unconjugated bilirubin,UCB),高浓度的UCB具有细胞毒性,可对神经系统造成不可逆损害[1]。(2)胆红素的运输:UCB难溶于水,为脂溶性物质,极易透过生物膜,与白蛋白有很强的亲和力。生理情况下,UCB可自由透过巨噬细胞的细胞膜进入血液,与血浆白蛋白形成复合体,从单核-巨噬细胞经血液循环运输至肝脏。到达肝脏后,UCB白蛋白复合物通过肝血窦内皮细胞的间隙进入窦周隙(Disse腔),在Disse腔中,UCB与白蛋白分离。分离后的UCB在肝细胞基底膜迅速被肝细胞摄取,该过程被认为是肝细胞基底外侧膜的有机阴离子转运多肽(organic anion transport polypeptide,OATP)1B1和OATP1B3发挥了作用[2]。进入肝细胞的UCB与肝细胞浆内的阴离子结合蛋白结合,首先是Y蛋白,当Y蛋白结合量接近饱和时,再与Z蛋白结合。此时,其水溶性增加,不易再反流入血,随后胆红素-载体蛋白复合物被运输至肝细胞滑面内质网的微粒体。(3)胆红素的酯化:在滑面内质网上,胆红素受尿苷二磷酸葡萄糖醛酸转移酶(uridine diphosphoglucuronate glucuronosyltransferase,UDPGT)催化,与葡萄糖醛酸进行1~2次结合反应,形成胆红素单葡萄糖醛酸酯(bilirubin monoglucuronide,BMG)和胆红素双葡萄糖醛酸酯(bilirubin diglucuronide,BDG),此过程是胆红素代谢的限速步骤。此外,小部分UCB在硫酸转移酶的作用下代谢为胆红素硫酸酯。上述双葡萄糖醛酸胆红素、单葡萄糖醛酸胆红素和硫酸胆红素,统称为结合胆红素(conjungated bilirubin,CB)。成人体内的CB约80%为BDG,而新生儿体内以BMG为主[3]。结合胆红素为水溶性,可通过肾小球滤过作用从尿中排出。(4)胆红素的排泄、重吸收及肠肝循环:胆红素主要经胆汁进行排泄,胆汁的主要成分是胆汁酸盐、胆红素和胆固醇。胆汁从肝细胞经胆小管转运蛋白分泌至毛细胆管,该过程是依赖ATP的逆浓度梯度转运。4种类型的胆小管转运蛋白中,微管多特异性有机阴离子转运体(canalicular multispecific organic anion transporter,cMOAT),又称为多药耐药蛋白2(multidrug resistance protein 2,MRP2)或ATP结合盒(ATP-binding cassette,ABC)C2,对胆红素分泌和除胆汁酸外的其他有机阴离子转运最为重要[4]。肝细胞所分泌的胆汁经毛细胆管微突、微胆管、细胆管、小胆管、肝总管、胆总管,经十二指肠乳头排入十二指肠。结合胆红素经胆道排入肠道后并不能被肠黏膜吸收,而是在回肠末端及结肠经厌氧菌还原酶作用还原为无色的胆素原,胆素原的大部分进一步氧化成为胆素,从粪便中排出体外,也称粪胆素。胆素原的小部分(约10%~15%)被回肠和结肠黏膜吸收,经门静脉血流回到肝内,大部分再经肝细胞作用后又转变为结合胆红素,又随胆汁排入肠道内,这一过程称为"胆红素的肠肝循环" 。小部分未能转变为结合胆红素,而是经血液循环进入肾脏,随尿排出体外,正常人每天由尿液排出的胆素原约0.5~4 mg。见图1。
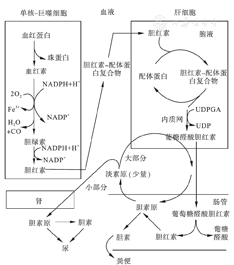
UDPGT:尿苷二磷酸葡萄糖醛酸转移酶;UDP:尿苷二磷酸。
胆红素的酯化是胆红素代谢的限速步骤,诱导UGT1A1基因表达可以提高酶活性从而促进机体的胆红素代谢。糖皮质激素受体(GR)、组成型雄甾烷受体(CAR)、孕烷X受体(PXR)等核受体可以通过调节UGT1A1的苯巴比妥类反应增强元件(PBREM)促进UGT1A1的转录表达。所以,苯巴比妥通过激活CAR、利福平激活PXR、地塞米松通过GR,都能发挥一定的降低胆红素水平的效果[5]。某些中草药含有上述核受体的激活成分,有潜在的药物开发价值。相反,某些抗肿瘤药物[6],例如伊立替康、埃罗替尼,抗HIV药物[7],例如阿扎那韦、茚地那韦,可抑制UGT1A1介导的胆红素结合反应,存在引起机体UCB浓度升高,导致黄疸的风险,尤其是存在UGT1A1基因突变者,风险更高。
此外,还有其他诸多因素都可影响胆红素代谢,例如血清白蛋白水平,临床显著的低蛋白血症势必影响UCB的运输,补充白蛋白有助于结合更多的游离胆红素,减少胆红素脑病的风险。头孢曲松是临床常用的三代头孢制剂,体外研究显示头孢曲松能取代胆红素与血清白蛋白结合,导致胆红素脑病的发生风险增高,因此,头孢曲松不得用于新生儿高胆红素血症者。
胆红素的产生过多、肝细胞对其摄取/酯化功能障碍,以及排泄减少等均可导致血液中胆红素水平升高,当血清中总胆红素浓度超过34.2 μmol/L时,可以观察到巩膜及皮肤的黄染,称为黄疸;若血清中胆红素浓度在正常以上,但不超过34.2 μmol/L,肉眼观察不到皮肤巩膜黄染,称隐性黄疸。临床根据其胆红素代谢异常所发生的环节,将黄疸分为以下3类:
因胆红素产生过多,超过肝脏的摄取、转化和排泄能力所导致的黄疸。胆红素的主要来源为血红蛋白,所以各种原因的血红蛋白破坏(即溶血)是肝前性黄疸最常见的原因。例如新生儿溶血、异型输血后溶血、自身免疫性溶血性贫血、遗传性葡萄糖-6-磷酸脱氢酶缺乏(蚕豆病)、遗传性球形红细胞增多症、阵发性睡眠性血红蛋白尿、恶性疟疾、伯氨奎林等药物中毒、蛇毒/毒蕈中毒等。肝前性黄疸主要表现为血清总胆红素增高,其中以UCB为主。
肝脏是胆红素代谢的关键器官,先天性基因缺陷或各种原因使肝脏受损导致的肝细胞摄取、酯化和排泄胆红素功能障碍,均可引起血清胆红素的增高。酯化功能障碍者以UCB升高为主,而胆红素排泄障碍则表现为CB升高,继发于肝损伤者则会出现UCB和CB的双向升高。
又称阻塞性黄疸,是由于胆道不通畅,导致肝内转化生成的结合胆红素从胆道系统排出困难,而反流入血,出现黄疸。常见于先天性胆道闭锁、各种遗传性肝病、胆管炎症、肿瘤等疾病。其特征为血中结合胆红素增高,总胆红素升高,结合胆红素占总胆红素的50%以上。
CNS是UGT1A1基因变异导致体内尿苷二磷酸葡萄糖醛酸转移酶(UDPGT)缺乏或活性低下引发的先天性胆红素结合功能障碍性疾病,是一种常染色体隐性遗传病。1952年首先被Crigler和Najjar[8]报道,多见于新生儿,但其发病率极低,在100万新生儿中约有1例。1962年Arias[9]提出CNS实际上根据酶活性可分为两型,CNS-Ⅰ型的酶完全缺乏,血清总胆红素波动于20~45 mg/dl,往往在新生儿期就出现严重的、持续性黄疸,有些患儿在生后数周或数月死于核黄疸,幸存者可伴或不伴神经损伤。CNS-Ⅱ型由于酶活性尚存一部分(通常为正常酶活性的10%),黄疸较Ⅰ型轻,血清总胆红素波动于6~20 mg/dl,但在饥饿、疾病状态下也可升至40 mg/dl。仅一半左右的患儿在1岁以内出现黄疸,通常可以存活至成年,不伴神经系统和智力的损伤,但之后仍可缓慢进展为胆红素脑病。
UGT1A1基因位于2号染色体长臂37区(2q37),由5个外显子组成,缺失、插入、错义突变、提前出现终止密码子等编码序列的变异均可导致其编码的尿苷二磷酸葡萄糖醛酸转移酶活性部分或完全丧失[10]。CNS-Ⅰ型中,基因损伤的位置常为编码信号肽的区域或酶的其他结构域异常,或是剪切体或受体区域的内含子序列异常,而导致编码提前终止或关键氨基酸序列的缺失。CNS-Ⅱ型中,常由点突变导致单个氨基酸的替换,使得酶活性降低但不至于完全缺乏。苯巴比妥可诱导CNS-Ⅱ型中尚存的UGT1A1酶活性,而对CNS-Ⅰ型无效。既往的CNS-Ⅰ型患儿往往在生后早期因核黄疸夭折,目前肝移植被认为是能使CNS-Ⅰ型患儿获得较长生存期的唯一确定有效的治疗方法[11],鉴于胆红素脑病的不可逆性,有学者呼吁尽可能早期进行预防性肝移植治疗。期待未来的基因治疗给CNS-Ⅰ型患儿带来新曙光[12]。
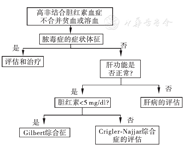
GS是UGT1A1基因变异导致体内UDPGT活性偏低的一种良性先天性高非结合胆红素血症。GS中UGT1A1基因异常包括两种类型:(1)启动子区域TATA框插入型突变,为常染色体隐性遗传,在TATA框中有TA插入,使正常A(TA)6TAA突变为A (TA)7TAA,此突变在西方人中常见[13];(2)外显子区域单碱基突变,为常染色体显性遗传,东方人中多为此类型,其中,第1外显子(Gly71Arg)较为多见,另外,第1外显子(Pro229Gln)、第4外显子(Arg367Gly)、第5外显子(Tyr486Asp)的突变等也有报道[14]。正常人群的UGT1A1基因变异频率较高(西方人群中约51%拥有至少一个Gilbert类型的等位基因),因此不同人群中GS患病率约为4%~16%。与CNS不同,GS患者的酶活性仅轻度受损,血清胆红素通常波动于1~6 mg/dl,其黄疸发作可由导致胆红素生成增加的状况触发,如禁食、溶血、感染、应激、体力活动、月经期等,较少诊断于青春期前,大多无需特殊治疗。新生儿期合并ABO溶血或G6PD酶缺乏症时也可被发现[15]。亦有观点认为CNS-Ⅰ型、CNS-Ⅱ型、GS均可归为UGT1A1异常谱系疾病[16,17]。临床上发现高非结合胆红素血症的患者,如果不存在贫血及溶血问题,可按图2流程进行鉴别诊断。
最初报道于上世纪60年代[18],又称暂时性新生儿家族性高胆红素血症,其病因被认为是孕妇妊娠后期血液循环及患儿血液中含有较高浓度的葡萄糖醛酸转移酶抑制剂,患儿可出现未结合胆红素明显增高,甚至发生核黄疸。但后续病例少,具体抑制剂的种类有待界定[17]。
DJS是一种因MRP2/ABCC2基因缺陷导致的先天性胆红素排泄障碍性疾病,为常染色体隐性遗传病[17]。1954年由Dubin和Johnson首先报道,呈现以结合胆红素为主的慢性间歇性黄疸发作[19]。该病较为罕见,发病率仅在犹太人群中略高,约为1∶3 000。MRP2/ABCC2基因位于10号染色体长臂24区(10q24),包含32个外显子。外显子跳读、错义突变、无义突变、碱基缺失都可导致转运蛋白表达受损。大多数突变导致的是终止密码出现和MRP2/ABCC2转录错误。较少出现的是MRP2/ABCC2的内质网滞留和无法转运至微管膜[20,21]。MRP2/ABCC2基因缺陷,胆小管转运蛋白功能受损,结合胆红素排泄受阻,出现高结合胆红素血症。由于结合胆红素在血浆中浓度升高后,可经肾脏排泄,故结合胆红素升高水平不重。DJS多于青年时期发现,通常表现为轻度的高结合胆红素血症,血清胆红素一般处于2~5 mg/dl,也可降至正常或在疾病、妊娠等状态下升至20-25 mg/dl。除此之外,患者一般无症状,也可合并腹部隐痛、乏力等全身症状。由于胆汁酸排泄无异常,患者不会出现瘙痒症状。除黄疸外,体格检查通常正常,偶尔可见肝脾肿大,但不会进展为纤维化或肝硬化。DJS极少数情况下可在新生儿期发病,可伴有严重的胆汁淤积和巨脾,胆红素水平可>20 mg/dl。新生儿期起病者症状严重可能和胆管生理功能尚未发育成熟有关。随着肝脏发育成熟,患儿症状可消失,仅表现为间歇性的高胆红素血症[22]。DJS预后较好,一般不需治疗。
RS是一种良性的、因OATP1B1和OATP1B3基因缺陷导致先天性胆红素再摄取及储存功能障碍性疾病。RS患者肝细胞排泄胆红素至毛细胆管的功能正常,但对肝细胞摄取肝血窦中的胆红素及胆红素的储存出现障碍。为常染色体隐性遗传病,是一种罕见病。SLCO1B1、SLCO1B3基因位于12p12.1和12p12.2,分别编码有机阴离子转运蛋白OATP1B1和OATP1B3。一般认为,OATP1B1和OATP1B3在功能上有重叠,当SLCO1B1、SLCO1B3基因均发生失活突变或缺失时方致病[23]。胆红素从肝细胞跨胆小管膜分泌至毛细胆管是胆红素排泄的限速步骤,可能出现过饱和现象。因此,另有一部分胆红素通过肝细胞血窦面的ATP-水解耦联泵ABCC3转运回肝血窦中。肝血窦下游靠近门静脉和肝动脉入口处的OATPB1和OATPB3对这些结合胆红素进行再摄取,从而将其转运到更多的肝细胞以增加肝脏排泄胆红素的能力。当OATPB1和OATPB3功能受损时,胆红素再摄取过程受阻。同时,RS患者肝脏对结合胆红素的储存能力也有缺陷,胆红素会渗漏入血浆,表现出高胆红素血症。RS患者肝脏储存能力缺陷的分子机制还有待进一步研究。RS表现为慢性的以结合胆红素升高为主的高胆红素血症,不伴溶血。血清总胆红素一般为2~5 mg/dl,也可更高。谷丙转氨酶、谷草转氨酶、碱性磷酸酶、γ-谷氨酰转肽酶正常。患者除黄疸外,一般无其他症状。该病为良性疾病,无需特殊治疗。
新生儿高胆红素血症,又称新生儿黄疸是一个笼统的概念,由于生命初期,胆红素的产生增多,但肝脏的摄取、转化及排泄功能尚不成熟或存在缺陷,一定比例的新生儿出现胆红素明显增高的现象,极度增高者可能出现核黄疸,甚至危及生命。新生儿黄疸的评估需结合胎龄、日龄、有无危险因素(不良家族史、新生儿窒息、败血症、低蛋白血症、代谢性酸中毒、溶血、巨大头颅血肿等)。我国及全球不同地区均制定了针对新生儿黄疸的专家共识[24],其中提供了较为详细的诊治建议供临床医师参考。
母乳性黄疸是新生儿黄疸中较常见的种类,见于纯母乳喂养或以母乳喂养为主者。既往认为其与母乳中的孕二醇等成分对葡萄糖醛酸转移酶有抑制作用,以及胆红素的肠-肝循环增加有关。近年研究显示母乳性黄疸婴儿存在UGT1A1的变异[25]。新生儿的生长发育多良好,需除外其他非生理性高胆红素血症。我国专家共识[24]建议总胆红素<15 mg/dl不需要停母乳;>15 mg/dl时可暂停母乳3 d,改人工喂养;>20 mg/dl则加用光疗。母乳喂养性黄疸见于新生儿最初3~5 d,与母乳喂养不足、胎粪排出延迟者有关,常有生理性体重下降>12%。根据专家共识[24]中的标准决定是否需要干预,应帮助母亲建立成功的母乳喂养,必要时补充配方乳。
综上,胆红素代谢是一个多步骤的复杂过程,环节及影响因素多,抓住胆红素酯化这一关键步骤,理解其上下游的代谢过程,有助于把握相关疾病的机制与相应临床表现。随着生命科学的发展,胆红素代谢的分子生物学机制被更多地揭示,本文介绍的几种疾病主要聚焦于"狭义"的胆红素代谢异常[26],希望能给大家临床诊治工作提供参考。
所有作者均声明不存在利益冲突

























