
探讨骨髓增生异常综合征(MDS)RAS基因突变的分子学特征及其对预后的影响。
收集2011年12月至2018年12月新诊断且完成二代测序的776例MDS患者病例资料,回顾性分析RAS基因突变患者的临床及分子学特征,并比较不同突变状态对总生存(OS)的影响。
共52例(6.7%)患者伴有RAS基因突变,NRAS突变38例(4.9%),KRAS突变18例(2.3%),4例(0.5%)同时伴NRAS及KRAS突变,全部NRAS突变及65%的KRAS突变位于第12、13及61号密码子。RAS基因突变与PTPN11、FLT3、U2AF1、RUNX1、WT1、ETV6及NPM1突变呈显著正相关(Q<0.05),且常为亚克隆突变。伴RAS基因突变的患者中诊断为MDS伴原始细胞增多(MDS-EB)的比例(82.7%)明显高于无RAS突变患者(35.2%)(P<0.001)。与无RAS基因突变患者相比,伴RAS基因突变患者的外周血白细胞水平和中性粒细胞水平明显升高[WBC:4.33(0.98~20.42)×109/L对2.71(0.61~21.17)×109/L,P<0.001;ANC:2.13(0.18~17.37)×109/L对1.12(0~16.00)×109/L,P<0.001],血小板水平明显减低[48(2~430)×109/L对62(2~694)×109/L,P=0.048],骨髓粒系比例有升高的趋势(47%对40%,P=0.085),骨髓原始细胞比例明显升高(7%对2%,P<0.001)。按照修订的国际预后积分系统(IPSS-R)进行预后分层,RAS基因突变患者中较高危分组(高危和极高危组)的比例显著增高(71.1%对37.9%,P<0.001)。单因素分析中,NRAS突变与更短的OS显著相关(P=0.011),经多因素校正后,NRAS突变对OS的影响失去显著性,突变基因中仅PTPN11及SETBP1为独立的不良OS因素。
RAS基因突变常作为亚克隆发生在MDS疾病晚期且常与转录因子及信号转导相关的突变伴随发生。RAS通路相关的PTPN11突变在MDS中为独立不良预后因素。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
RAS信号通路在调节细胞增殖、分化和凋亡的过程中发挥重要作用,RAS蛋白的持续激活可以导致信号传导紊乱,促进细胞的增殖及恶性转变[1]。既往研究表明RAS癌基因的激活突变常见于多种血液系统恶性肿瘤,包括急性髓系白血病(AML)、骨髓增生异常综合征(MDS)、骨髓增生异常综合征/骨髓增殖性肿瘤(MDS/MPN)等,且常与疾病进展相关[2,3,4]。我们采用二代测序在776例初诊原发性MDS患者中发现52例伴有RAS基因突变,本文对RAS基因突变患者的分子学、临床和实验室特征,及其对总生存(OS)的影响进行了分析,现报道如下。
2011年12月至2018年12月于中国医学科学院血液病医院MDS和MPN诊疗中心确诊并完成二代测序的初诊MDS患者纳入研究,共776例,男494例(63.7%),中位年龄54(14~83)岁。按照《骨髓增生异常综合征中国诊断与治疗指南(2019年版)》标准[5],MDS伴单系发育异常(MDS-SLD)34例(4.4%),MDS伴多系发育异常(MDS-MLD)378例(48.7%),MDS伴环形铁粒幼红细胞(MDS-RS)37例(4.7%),MDS伴原始细胞增多-1(MDS-EB-1)153例(19.7%),MDS伴原始细胞增多-2(MDS-EB-2)145例(18.7%),MDS伴5q- 7例(0.9%),MDS未分类(MDS-U)22例(2.8%)。678例(87.4%)患者具有可分析的染色体核型结果,采用修订的国际预后积分系统(IPSS-R)[6]对患者进行预后分层:极低危组21例(3.1%)、低危组169例(24.9%)、中危组216例(31.9%)、高危组152例(22.4%)、极高危组120例(17.7%)。575例患者可追踪到治疗方案,其中48例(8.3%)接受单纯支持治疗,303例(52.7%)接受免疫抑制剂或免疫调节剂治疗,91例(15.8%)接受地西他滨治疗,74例(12.9%)接受造血干细胞移植,38例(6.6%)接受CAG/HAG(阿克拉霉素/高三尖杉酯碱+阿糖胞苷+G-CSF)方案化疗,21例(3.7%)接受"中医"治疗。
骨髓细胞经过24 h培养,收集细胞常规制片,R显带,根据《人类细胞遗传学国际命名体制(ISCN2013)》描述核型异常。按照IPSS-R染色体核型分组标准[6]进行染色体核型预后分组。
所有患者DNA样本来源于骨髓单个核细胞。使用PCR引物扩增目的基因组(涵盖112个血液肿瘤相关基因),富集后采用Ion Torrent半导体测序平台进行测序。平均基因覆盖率98.1%,平均测序深度1310×。测序后原始数据利用CCDS、人类基因组数据库(HG19)、dbSNP(v138)、1000genomes、COSMIC、PolyPhen-2等数据库进行生物信息学分析,筛选致病性基因突变位点。具体方法参见本研究组此前已发表文献[7]。
采用等位基因突变频率(VAF)对携带两种及以上突变的患者进行突变时序分析。参考文献[8,9]方法,VAF值显著高的突变被判定为主克隆,显著低的突变被判定为亚克隆,当不同突变的VAF值之间无显著差异时,其均被判定为主克隆。
随访截止时间为2019年5月31日,随访资料来源于住院病历、门诊病历及电话随访记录。对随访期间死亡的病例,按照病历记录或与患者家属电话联系确认。中位随访时间为17(2~87)个月,失访患者共89例(11.5%)。OS期指自诊断日期到死亡或末次随访日期。
统计分析利用SPSS 25.0及R 3.6.1软件完成。非正态分布的计量资料以"中位数(范围)"表示,采用Mann-WhitneyU检验进行组间比较;分类变量组间比较采用卡方检验或Fisher精确概率法,并采用Benjamini-Hochberg法对双侧检验P值进行多重校正,Q<0.05为差异有统计学意义。突变时序分析采用皮尔森拟合优度检验[9]。采用Kaplan-Meier法绘制生存曲线,Log-rank检验进行单因素分析,Cox等比例回归进行多因素分析,双侧检验P<0.05为差异有统计学意义。应用Graphpad Prism 7.0及R 3.6.1绘图。
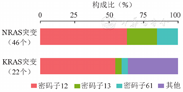
776例MDS患者中共发现NRAS和(或)KRAS突变52例(6.7%),其中NRAS突变38例(4.9%),KRAS突变18例(2.3%),4例(0.5%)患者同时伴有NRAS及KRAS突变。52例患者共检出46个NRAS突变及22个KRAS突变,皆为点突变。全部NRAS突变及65%的KRAS突变均发生在第12、13和61号密码子,其中以第12号密码子最为常见,分别占NRAS突变的63%和KRAS突变的55%(图1)。
伴有RAS突变的患者携带其他基因的平均突变数为(3.71±1.46)个,其中NRAS突变患者平均为(3.68±1.44)个,KRAS突变患者平均为(4.06±1.51)个,高于无RAS基因突变患者的(1.76±1.44)个。除NRAS/KRAS突变外,全部776例患者中突变频率较高(>5%)的基因还有U2AF1(20.4%)、ASXL1(12.4%)、SF3B1(9.4%)、TP53(7.3%)、RUNX1(7.1%)、DNMT3A(6.7%)、TET2(6.3%)和SETBP1(5.8%)。
RAS突变常与PTPN11(Q<0.001,OR=7.096)、FLT3(Q<0.001,OR=18.945)、U2AF1(Q=0.012,OR=2.206)、RUNX1(Q=0.025,OR=2.610)、WT1(Q=0.006,OR=7.416)、ETV6(Q=0.039,OR=9.534)及NPM1(Q=0.012,OR=4.409)突变同时发生(图2)。NRAS突变分别与KRAS、PTPN11、FLT3、WT1及NPM1突变呈正相关(Q值均<0.05),KRAS与U2AF1突变呈正相关(Q<0.05)(图2)。
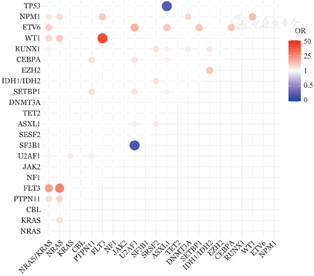
红色为呈正相关,蓝色为呈负相关;Q值以圆圈大小表示,Q值越小,圆圈越大
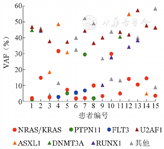
RAS突变的中位VAF为7.30(1.40~49.07),其中NRAS突变为7.85(1.77~49.07),KRAS突变为4.90(1.40~48.34)。各诊断分型及预后分组之间RAS突变VAF无显著差异(P值均>0.05)。对39例出现1个及以上除NRAS和(或)KRAS以外基因突变的患者的VAF进行分析,其中8例(20.5%)患者的NRAS/KRAS突变为主克隆,31例(79.5%)为亚克隆,NRAS突变和KRAS突变中亚克隆的比例分别为75.0%(21/28)和85.7%(12/14),提示RAS基因突变在MDS中常常为发生于晚阶段的分子事件。图3展示了15例除NRAS/KRAS以外2个以上基因突变的患者各基因VAF的分布情况。
我们分别将RAS突变组、NRAS突变组及KRAS突变组患者的临床及实验室特征与无RAS突变组患者进行比较。各组之间性别与年龄分布差异均无统计学意义(P值均>0.05)。根据WHO(2016)诊断及分型标准,RAS突变组、NRAS突变组和KRAS突变组中MDS-EB的比例(分别为82.7%,81.6%和83.3%)均显著高于无RAS突变组(35.2%,P值均<0.001)。与无RAS突变组相比,三组RAS突变患者的外周血WBC和ANC水平均明显升高(P值均<0.05),RAS突变组PLT水平明显减低(P=0.048),各组之间HGB水平无明显差异(表1)。RAS突变组患者骨髓粒系比例较无RAS突变组有升高的趋势(P=0.085),三组伴RAS突变患者的骨髓原始细胞比例均明显高于无RAS突变患者(P值均<0.001)(表1)。
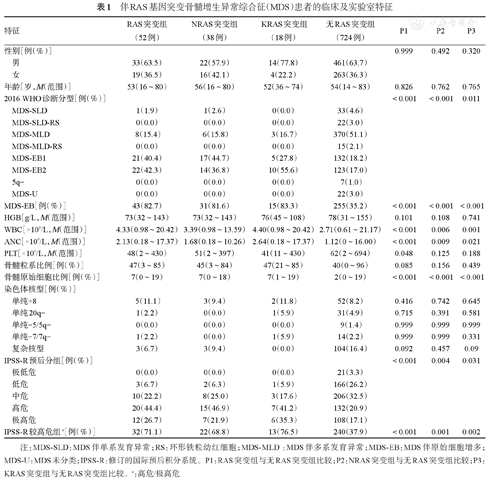
伴RAS基因突变骨髓增生异常综合征(MDS)患者的临床及实验室特征
伴RAS基因突变骨髓增生异常综合征(MDS)患者的临床及实验室特征
| 特征 | RAS突变组(52例) | NRAS突变组(38例) | KRAS突变组(18例) | 无RAS突变组(724例) | P1 | P2 | P3 | |
|---|---|---|---|---|---|---|---|---|
| 性别[例(%)] | 0.999 | 0.492 | 0.320 | |||||
| 男 | 33(63.5) | 22(57.9) | 14(77.8) | 461(63.7) | ||||
| 女 | 19(36.5) | 16(42.1) | 4(22.2) | 263(36.3) | ||||
| 年龄[岁,M(范围)] | 53(16~80) | 56(16~80) | 52(36~74) | 54(14~83) | 0.826 | 0.762 | 0.765 | |
| 2016 WHO诊断分型[例(%)] | <0.001 | <0.001 | 0.011 | |||||
| MDS-SLD | 1(1.9) | 1(2.6) | 0(0.0) | 33(4.6) | ||||
| MDS-SLD-RS | 0(0.0) | 0(0.0) | 0(0.0) | 22(3.0) | ||||
| MDS-MLD | 8(15.4) | 6(15.8) | 3(16.7) | 370(51.1) | ||||
| MDS-MLD-RS | 0(0.0) | 0(0.0) | 0(0.0) | 15(2.1) | ||||
| MDS-EB1 | 21(40.4) | 17(44.7) | 5(27.8) | 132(18.2) | ||||
| MDS-EB2 | 22(42.3) | 14(36.8) | 10(55.6) | 123(17.0) | ||||
| 5q- | 0(0.0) | 0(0.0) | 0(0.0) | 7(1.0) | ||||
| MDS-U | 0(0.0) | 0(0.0) | 0(0.0) | 22(3.0) | ||||
| MDS-EB[例(%)] | 43(82.7) | 31(81.6) | 15(83.3) | 255(35.2) | <0.001 | <0.001 | <0.001 | |
| HGB[g/L,M(范围)] | 73(32~143) | 73(32~143) | 76(45~108) | 78(31~155) | 0.101 | 0.108 | 0.741 | |
| WBC[×109/L,M(范围)] | 4.33(0.98~20.42) | 3.39(0.98~13.59) | 4.40(0.98~20.42) | 2.71(0.61~21.17) | <0.001 | 0.006 | 0.001 | |
| ANC[×109/L,M(范围)] | 2.13(0.18~17.37) | 1.68(0.18~10.26) | 2.64(0.18~17.37) | 1.12(0~16.00) | <0.001 | 0.009 | 0.021 | |
| PLT[×109/L,M(范围)] | 48(2~430) | 51(2~397) | 41(11~430) | 62(2~694) | 0.048 | 0.125 | 0.188 | |
| 骨髓粒系比例[例(%)] | 47(3~85) | 45(3~84) | 47(21~85) | 40(0~96) | 0.085 | 0.156 | 0.439 | |
| 骨髓原始细胞比例[例(%)] | 7(0~19) | 7(0~18) | 7(1~19) | 2(0~19) | <0.001 | <0.001 | <0.001 | |
| 染色体核型[例(%)] | ||||||||
| 单纯+8 | 5(11.1) | 3(9.4) | 2(11.8) | 52(8.2) | 0.416 | 0.742 | 0.645 | |
| 单纯20q- | 1(2.2) | 0(0.0) | 1(5.9) | 31(4.9) | 0.715 | 0.391 | 0.581 | |
| 单纯-5/5q- | 0(0.0) | 0(0.0) | 0(0.0) | 9(1.4) | 0.999 | 0.999 | 0.999 | |
| 单纯-7/7q- | 1(2.2) | 0(0.0) | 1(5.9) | 14(2.2) | 0.999 | 0.999 | 0.331 | |
| 复杂核型 | 3(6.7) | 3(9.4) | 0(0.0) | 104(16.4) | 0.092 | 0.457 | 0.09 | |
| IPSS-R预后分组[例(%)] | <0.001 | 0.004 | 0.031 | |||||
| 极低危 | 0(0.0) | 0(0.0) | 0(0.0) | 21(3.3) | ||||
| 低危 | 3(6.7) | 2(6.3) | 1(5.9) | 166(26.2) | ||||
| 中危 | 10(22.2) | 8(25.0) | 3(17.6) | 206(32.5) | ||||
| 高危 | 20(44.4) | 15(46.9) | 7(41.2) | 132(20.9) | ||||
| 极高危 | 12(26.7) | 7(21.9) | 6(35.3) | 108(17.1) | ||||
| IPSS-R较高危组a[例(%)] | 32(71.1) | 22(68.8) | 13(76.5) | 240(37.9) | <0.001 | 0.001 | 0.002 | |
注:MDS-SLD:MDS伴单系发育异常;RS:环形铁粒幼红细胞;MDS-MLD :MDS伴多系发育异常;MDS-EB:MDS伴原始细胞增多;MDS-U:MDS未分类;IPSS-R:修订的国际预后积分系统。P1:RAS突变组与无RAS突变组比较;P2:NRAS突变组与无RAS突变组比较;P3:KRAS突变组与无RAS突变组比较。a:高危/极高危
RAS突变与各染色体核型之间无显著相关性。按照IPSS-R对患者进行预后分组,RAS突变组、NRAS突变组及KRAS突变组中较高危分组(IPSS-R高危和极高危组)所占的比例均显著高于无RAS突变组(P值均<0.05)。各组之间临床及实验室特征的比较详见表1。
截止末次随访,随访患者中死亡228例(33.1%),其中RAS突变组23例(48.9%),NRAS突变组18例(50.0%),KRAS突变组5例(35.7%),无RAS突变组205例(32.0%)。中位OS期:RAS突变组患者为21(95%CI 12.72~29.29)个月,NRAS突变组为21(95%CI 4.58~37.42)个月,KRAS突变组未达到,无RAS突变组为52(95%CI 32.47~71.53)个月。
单因素分析中,伴有RAS及NRAS基因突变患者的OS期较无突变患者显著缩短(P=0.003、0.011),而KRAS突变对OS无显著影响(P=0.491)(图4A,图4B,图4C)。RAS、NRAS及KRAS的主克隆和亚克隆突变患者之间的OS差异无统计学意义(P值均>0.05)(图4D,图4E,图4F)。突变频率>2%的其他基因突变中,还有TET2、SETBP1、PTPN11和TP53突变与更短的OS期显著相关(P值均<0.05),SRSF2和RUNX1突变患者的OS期有缩短的趋势(P=0.090、0.084)(表2)。将单因素分析中P<0.1的基因以及年龄(<60岁/≥60岁)、IPSS-R预后分层纳入COX等比例风险模型,NRAS基因突变对OS的影响失去显著性(P=0.825),而PTPN11(HR=2.137,95% CI 1.068~4.278,P=0.032)、SETBP1(HR=1.936,95% CI 1.089~3.442,P=0.024)为OS的独立预后不良因素(表2)。
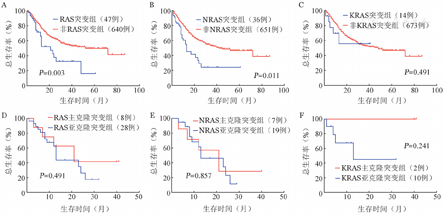
A:RAS突变;B:NRAS突变;C:KRAS突变;D:RAS突变时序;E:NRAS突变时序;F:KRAS突变时序
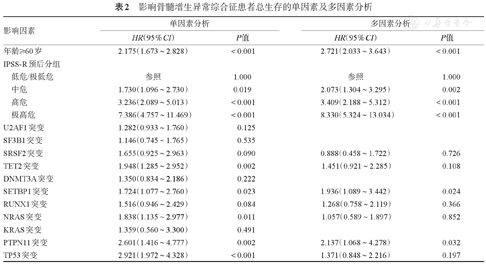
影响骨髓增生异常综合征患者总生存的单因素及多因素分析
影响骨髓增生异常综合征患者总生存的单因素及多因素分析
| 影响因素 | 单因素分析 | 多因素分析 | ||||
|---|---|---|---|---|---|---|
| HR(95%CI) | P值 | HR(95%CI) | P值 | |||
| 年龄≥60岁 | 2.175(1.673~2.828) | <0.001 | 2.721(2.033~3.643) | <0.001 | ||
| IPSS-R预后分组 | ||||||
| 低危/极低危 | 参照 | 1.000 | 参照 | 1.000 | ||
| 中危 | 1.730(1.096~2.730) | 0.019 | 2.073(1.304~3.295) | 0.002 | ||
| 高危 | 3.236(2.089~5.013) | <0.001 | 3.409(2.188~5.312) | <0.001 | ||
| 极高危 | 7.386(4.757~11.469) | <0.001 | 8.330(5.324~13.034) | <0.001 | ||
| U2AF1突变 | 1.282(0.933~1.760) | 0.125 | ||||
| SF3B1突变 | 1.146(0.745~1.765) | 0.535 | ||||
| SRSF2突变 | 1.655(0.925~2.963) | 0.090 | 0.888(0.458~1.722) | 0.726 | ||
| TET2突变 | 1.948(1.285~2.952) | 0.002 | 1.451(0.921~2.285) | 0.108 | ||
| DNMT3A突变 | 1.350(0.834~2.186) | 0.222 | ||||
| SETBP1突变 | 1.724(1.077~2.760) | 0.023 | 1.936(1.089~3.442) | 0.024 | ||
| RUNX1突变 | 1.516(0.946~2.429) | 0.084 | 1.268(0.758~2.119) | 0.366 | ||
| NRAS突变 | 1.838(1.135~2.977) | 0.011 | 1.057(0.589~1.897) | 0.852 | ||
| KRAS突变 | 1.359(0.560~3.300) | 0.491 | ||||
| PTPN11突变 | 2.601(1.416~4.777) | 0.002 | 2.137(1.068~4.278) | 0.032 | ||
| TP53突变 | 2.921(1.972~4.328) | <0.001 | 1.371(0.848~2.216) | 0.197 | ||
RAS基因家族由NRAS、KRAS和HRAS三个成员组成。其编码的RAS蛋白经翻译后修饰可定位于细胞膜内侧并通过RAS/RAF/MEK和RAS/PI3K等信号通路传导细胞信号,并最终激活核内转录因子,调控细胞增殖、分化和凋亡[10]。RAS基因的激活突变可导致RAS通路的持续活化及细胞的过度增殖、凋亡减少,与髓系肿瘤的发生密切相关[1]。此外,编码RAS蛋白上游分子的基因(FLT3、CBL等)或参与RAS信号通路调节的基因(PTPN11、NF1等)发生突变均可导致RAS调控的紊乱[11]。
RAS突变为MDS中首个被发现的基因突变。二十世纪八九十年代,即有多个小样本研究报道了在MDS患者中检出RAS基因突变,这些基于寡核苷酸探针的杂交测序技术大多特异地针对第12、13和61号密码子的热点突变进行检测,检出率为3%~48%[12,13,14,15],并发现RAS突变常在特定的FAB分型中富集,包括难治性贫血伴原始细胞增多(RAEB)、难治性贫血伴原始细胞增多转变型(RAEB-t)、慢性粒-单核细胞白血病(CMML)[12,14,16]。一代测序技术的应用使得RAS突变的检出率较前有所下降,但仍差异较大,在0~25%之间[17,18],可能由于患者的构成差异、样本量的限制、不同的检测方法等多个因素所致。随着MDS诊断标准从FAB标准过渡到WHO诊断分型标准,将FAB分型中的RAEB-t归为AML,CMML归为MDS/MPN[19];随着高通量的二代测序技术的广泛应用,报道的MDS患者RAS基因突变频率逐渐趋于一致,在5%~10%之间,其中NRAS突变率约为5%,KRAS突变率为2%[20,21]。本研究队列中NRAS及KRAS基因的突变频率及位点分布情况与上述二代测序结果基本一致。
有研究表明,RAS基因突变与RAS通路相关的基因突变在AML、幼年型粒-单核细胞白血病(JMML)等多种髓系肿瘤中互斥,提示RAS通路中的任一基因单独发生突变即可导致RAS通路的调控紊乱从而获得生长增殖优势[22,23]。而在MDS中RAS突变与其他基因突变之间的相关性尚不十分明确。曾有研究报道RAS突变与RUNX1、ASXL1、EZH2、BCOR、STAG2等突变呈正相关,与SF3B1突变呈负相关[9,24]。有意思的是,我们的结果提示RAS突变与PTPN11、FLT3等RAS通路相关的基因突变呈正相关,且常与其他信号转导及转录因子相关基因突变同时发生,如RUNX1、NPM1、WT1等,这与Ogawa等[25]、Makishima等[26]的研究接近。我们推测突变相关性不一致可能的原因有:①不同检测方法的检出率及检测位点不一致;②不同队列中某些突变的发生频率不一致;③相比于AML、JMML等,RAS通路突变在MDS中发生更晚,突变负荷更低,发生单一突变的克隆获得增殖优势的能力较弱,使得携带不同突变的亚克隆共存,并加速疾病的进展。
除了相关性,我们还就RAS基因突变在MDS克隆演变中的发生阶段进行了探讨。结果表明,RAS突变多为亚克隆,其突变负荷明显低于U2AF1、DNMT3A、ASXL1等,并与PTPN11、FLT3等接近。近年来,随着测序技术的发展,研究者们对MDS突变谱系及克隆演进模式的认识逐步深入。几个大系列的研究均提示剪接子及DNA甲基化相关的突变多发生在疾病早期,为MDS的驱动突变;染色质修饰与转录因子相关的突变则发生相对较晚,却通常早于RAS通路相关基因突变[9,20,27]。Makishima等[26]发现相较于高危MDS,NRAS、FLT3、PTPN11等7个基因在继发性AML中高度富集;相比于低危MDS,KRAS、TP53、RUNX1等8个基因在高危MDS中聚集出现。以上结果均证明RAS基因突变发生于MDS的较晚阶段,并与疾病进展相关。
迄今所报道的RAS基因突变大多发生在较晚期的MDS,如RAEB、RAEBt、CMML[14,16,28],而在难治性贫血(RA)、环形铁粒幼细胞性难治性贫血(RAS)中罕见。连续性研究表明RAS突变与MDS的进展和转AML相关[18,29,30]。本研究的结果显示RAS突变在MDS-EB中的发生率明显高于其他分型,与文献[18,29,30]报道一致。本研究中RAS突变患者的WBC明显升高,PLT明显减低,骨髓原始细胞比例明显增高,这与Bjar等[31]、Al-kali等[3]的结果一致。曾有研究报道RAS突变与染色体+8异常相关[32],而在我们的队列中未得到验证,可能与不同队列的患者构成比不同有关。鉴于较高的原始细胞比例及较低的PLT,我们发现RAS突变与IPSS-R较高危分组显著相关。
目前关于RAS突变在MDS中的预后效力尚无定论。部分研究表明RAS突变或单独NRAS突变提示预后不良[15,29,33],Hasse等[34]的结果提示NRAS突变在MDS伴复杂核型的患者中为独立的不良预后因素;也有学者认为RAS或NRAS突变状态与OS无明显相关性[3,18,31]。多数研究未发现KRAS与不良的OS相关,而Hefarlach等[20]对944例患者进行分析后发现KRAS突变为OS的独立预后不良因素,而NRAS突变对OS无影响。我们的结果表明,RAS突变,尤其是NRAS突变在单因素分析中与更短的OS期显著相关,KRAS突变无影响,而在纳入IPSS-R预后分层以及数个基因突变的多因素分析中NRAS突变的影响失去显著性,提示RAS突变,尤其是NRAS突变的不良预后可能主要由于其和部分高危因素的相关性所导致,如更高的骨髓原始细胞比例,高危的IPSS-R预后分层,伴发的信号转导及转录因子相关基因突变等。
我们的结果还表明PTPN11和SETBP1为独立的不良预后因素。PTPN11属于RAS通路相关基因,其突变在JMML中为主要分子事件之一[35],在MDS中少见。关于PTPN11在MDS中的研究较少,比较明确的是其在原发AML中的突变频率明显高于MDS[25],且与MDS进展为AML密切相关。PTPN11的预后价值还有待多中心大样本的分析加以证实。SETBP1为表观遗传相关基因,Tefferi等[36]的研究提示SETBP1为OS的独立预后不良因素,与我们的结果相一致。我们前期研究表明TP53突变在经复杂核型校正后不再是一独立的不良预后因素[37],本研究在扩大样本量后与前期研究结果一致。
综上,本研究证实RAS基因突变多发生在MDS的晚期,且常与转录因子及信号转导相关的突变伴随发生,共同促进疾病进展和白血病转化。RAS通路相关的PTPN11突变是一个独立的不良预后因素,对MDS患者的预后判断有一定的指导价值。本研究尚存在一些不足,由于在随访中无法获得所有患者是否存在明确的AML转化信息,因此没有回答RAS突变对MDS转AML的影响,此外,本研究为单中心回顾性分析,结论仍需多中心前瞻性研究加以证实。

























