
分子遗传学改变是Lennox-Gastaut综合征的重要致病因素之一,突变类型多为新生的单核苷酸变异,也有拷贝数变异、染色体异常等。现有报道共涉及近40个相关致病基因,涵盖离子通道、突触传递、生长发育等多方面。本文深入综述了近20余年来Lennox-Gastaut综合征分子遗传学的研究。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Lennox-Gastaut综合征(Lennox-Gastaut syndrome,LGS)是一种严重的年龄依赖性癫痫性脑病(epileptic encephalopathy,EE),起病高峰3~5岁,占儿童癫痫的1%~10%[1]。三大核心特征包括:(1)以强直发作为主的多种难治性癫痫发作,如不典型失神、肌阵挛发作等;(2)不同程度的认知受损或智力障碍;(3)脑电图特征表现为双侧或全导联<3 Hz的棘慢波,以及睡眠中的阵发快波节律[2]。约75%的LGS患者可找到相关的症状学病因如缺血缺氧性脑病、皮质发育异常、外伤、感染,其余25%患者病因不明,被称为特发性LGS[2]。
分子遗传学改变与LGS密不可分,LGS的遗传学病因多表现为单核苷酸变异(single nucleotide variation,SNV),包括无义、错义突变等,以及小片段(<50 bp)的插入或缺失,或涉及重要基因的拷贝数变异(copy number variation,CNV)、染色体病,目前尚缺乏表观遗传学方面的报道。LGS作为一种散发的EE,新生(de novo)突变居多,多呈显性遗传,家系报道少见。相关致病基因较为多样,涵盖离子通道、突触传递、神经系统发育、mTOR信号通路等多方面。离子通道基因是经典的癫痫基因,近年来非离子通道基因如突触传递相关基因也被逐渐发现,有报道称约75% EE的新生突变与突触转运相关[3]。
本文综述了近20余年来的研究结果,将对在LGS患者中发现的相关分子遗传学改变进行分类描述。
编码电压门控钠离子通道的α1亚基。致病机制多为功能缺失突变(loss of function,LOF),造成GABA能神经元功能低下,引起癫痫发作。SCN1A基因是经典的EE基因,70%以上的Dravet综合征由SCN1A基因突变导致,其中95%是新生突变[4]。SCN1A基因突变也可导致LGS[4,5,6],携带SCN1A基因突变的LGS患者癫痫发作以肌阵挛为主,具有某些类似Dravet综合征的特点。治疗上,对于SCN1A基因突变导致的LOF突变,钠离子通道阻滞剂的治疗效果差[7]。
编码电压门控钠离子通道的α2亚基。致病机制同时包括LOF突变和功能获得突变(gain of function,GOF),GOF造成可兴奋神经元去极化阈值降低等,而LOF造成Nav1.2激活减少,可能通过影响抑制性神经元的功能增加神经系统兴奋性。研究显示SCN2A基因突变可导致约1%的EE,包括LGS[6,8]。针对不同的致病机制,离子通道阻滞剂的治疗效果不同[7]。
编码电压门控钠离子通道的α8亚基,致病机制主要是GOF突变。经统计SCN8A基因突变占EE的1%~1.4%[9],且与癫痫猝死相关。SCN8A基因相关EE合并多种癫痫发作,起病后精神运动发育迟滞甚至倒退,突出表现为认知下降、小脑萎缩和共济失调[9]。
编码钙离子通道的α1A亚基。CACNA1A基因是人类基因组前2%最不耐受突变的基因之一,不同位点的突变分别导致LOF或者GOF,导致疾病发生。该基因是EE的明确致病基因,可导致LGS,患者均表现为多种类型的癫痫发作、不同程度的精神运动发育迟滞和小脑损害[10]。
编码突触融合蛋白结合蛋白1,通过与突触融合蛋白1A(Stx1a)、SNARE受体的特异性联系,调节突触递质的释放。STXBP1基因相关脑病表现为严重的发育迟滞、智力障碍和癫痫发作,可表现为大田原综合征、婴儿痉挛症、LGS等。杂合子致病机制主要是单倍体剂量不足,变异的STXBP1蛋白稳定性差,不能与Stx1a结合,影响突触递质释放。相反,纯合变异型主要造成GOF,不对蛋白水平及突触递质释放造成明显影响,并且患者特征性表现为LGS[11]。
编码一种突触前发动蛋白。该基因突变选择性影响抑制性突触,导致囊泡的内吞作用受损,囊泡循环效率低下,抑制性突触的丛集性放电减少,引起癫痫发作。DNM1基因突变约占严重癫痫的2%,可导致婴儿痉挛症或LGS。典型的DNM1基因相关EE起病几乎全表现为婴儿痉挛症,约80%患者随年龄发展为LGS,具有重度智力障碍,并且起病前即可有较为明确的精神运动发育迟滞、肌张力障碍和失语等[12]。
编码γ-氨基丁酸A型受体的β3亚基,GABAA受体是神经系统主要的抑制性受体,主要在突触后膜表达,作用于神经系统发育和抑制性兴奋传导。基因突变造成抑制性神经传导异常,可表现为儿童失神癫痫、LGS、婴儿痉挛症等。GABRB3基因相关EE缺乏明确的、共同的表型特点,Daniel等指出携带GABRB3基因突变的EE患者可能具有口面部畸形[13]。GABRB3基因是LGS的明确致病基因,变异的β3亚基在细胞内滞留,胞膜表面受体表达减少,并协同影响γ亚基的功能,引起GABA诱发的峰值电流幅度降低,GABA能神经元的抑制功能减弱而发病[14]。类似的,LGS患者也可携带GABRG2基因突变[15]。
编码GABA转运体1(GAT-1),是脑内主要的GABA转运体之一,作用于突触对GABA递质的清除和再摄取。SLC6A1基因突变与肌阵挛失张力癫痫(MAE)、智力障碍相关疾病密切相关。有学者报道1例LGS患者携带SLC6A1基因的新生突变(c.700G>A p.G234S),变异的GAT-1功能异常且表达总量减少,影响对GABA递质的摄取和清除而致病[16]。GAT-1作为一种依赖钠离子、氯离子GABA转运体,与离子通道功能、突触传递功能均密切相关。
编码细胞内钠氢离子交换泵6,对于神经元树突和突触形成至关重要。SLC9A6基因突变可导致Christianson综合征,一种X连锁疾病,患者表现为失语、共济失调、小头畸形、过度运动、智力障碍和难治性癫痫。Ikeda等[17]报道2例携带SLC9A6基因突变的Christianson综合征患者,癫痫发作均表现为LGS,认为Christianson综合征患者的癫痫发作更倾向发展为LGS这一EE。
编码染色体结构域解螺旋酶DNA结合蛋白2,通过染色体重塑等参与基因表达的负调控。CHD2基因突变可以导致一系列神经系统发育障碍,表现为严重的EE如LGS,患者具有典型的肌阵挛发作和光诱发的特点[20]。
编码脑特异性转录抑制因子,参与神经细胞的增殖、分化、迁移定位和凋亡等,与端脑发育密切相关。FOXG-1基因功能异常,神经细胞成熟障碍,神经细胞耗竭。癫痫发作多由FOXG-1基因的LOF突变所致。携带该基因突变的LGS患者,具有小头畸形、精神运动发育迟滞、言语障碍及Rett综合征样表现。治疗方面,LGS具有明显的药物难治性,而携带FOXG-1基因突变的其他癫痫患者通常对离子通道阻滞剂治疗效果好[21]。
参与编码尿苷二磷酸N乙酰葡萄糖转移酶,在前头部皮质和海马区域神经元特异性表达。该基因敲除的小鼠异常突触形成增多,阻碍脑内正常神经网络的建立。临床主要表现为婴儿痉挛症,少数表现为LGS,患儿可同时伴有先天性糖基化障碍。值得注意的是,携带ALG13基因突变的婴儿痉挛症患者,大多为同一位点的突变所致(c.320A>Gp.N107S),几乎全为女性患者[22]。
属于WD重复蛋白家族。该基因突变将导致β-螺旋桨蛋白相关神经变性(BPAN),呈X连锁隐性遗传,核心临床特征包括严重的EE如婴儿痉挛症、LGS及运动系统症状等。BPAN患者头颅核磁特征表现为黑质和苍白球的铁沉积,T2及SWI序列可见黑质和苍白球区域的低信号,T1序列上可见黑质和大脑脚附近的条带状低信号周围环状高信号[23]。WDR45基因突变与LGS关系密切,有可能是LGS的致病基因之一。
GPR56基因编码一种G蛋白偶联受体,GPR56表达低下造成额、顶叶新皮质的发育不良;DCX基因作用于神经元迁移和皮质发育,两者均有罕见家系报道。Parrini等[24]报道3个家系共4例LGS患者合并GPR56基因突变,头颅核磁提示特征性双侧额顶叶皮层多发小脑回畸形(bilateral frontoparietal polymicrogyria,BFPP)。Lawrence等[25]报道1个家系中3例LGS患者均携带DCX基因的致病性突变(c.4G>A p.E2K),影像学特征表现为巨脑回畸形。当涉及皮质发育的基因如GPR56、LIS1、DCX基因等发生突变,造成皮质结构异常如无脑回、巨脑回,导致LGS发病。
基因突变表现为严重的EE,多1岁内起病,早期表现为大田原综合征、婴儿痉挛症,后期向LGS进展。癫痫发作主要表现为强直发作和癫痫样痉挛,合并神经发育异常、认知倒退等。携带CDKL基因突变可具有Rett综合征表现、脑萎缩、特殊面容等[6]。
TSC(tuberous sclerosis complex)基因与LGS:结节性硬化是一种常染色体显性遗传病,85%的结节性硬化患者携带TSC1或TSC2基因突变。TSC复合物是mTOR通路的负性调节因子。TSC基因突变导致mTOR通路功能异常,累及脑、肺、皮肤等多系统,引起癫痫发作。26%的TSC癫痫患者表现为LGS或类LGS表现,且大部分由婴儿痉挛症发展而来[26]。
CNV:指基因序列(>1 kb)的插入、缺失或重复,38%的LGS患者携带可能致病的一个或多个CNV,比率明显高于健康人群、癫痫人群[27]。目前在LGS患者中报道的考虑可能致病的de novo CNV有22q13.3del、2q23.1del、Xq28dup、2q24del、5p15del、14q23del、15q11dup、16p13.2dup等[27,28]。患者携带的CNV不一定致病,由表型正常的双亲遗传而来的CNV,相对于新生突变,更倾向于良性变异。5%~10%的正常人也携带>500 kb的CNV,在存在结构性脑发育异常的患者中,CNV的发生频率更高[27]。
1%~13%的Down综合征患者可出现各种类型的癫痫发作,其中婴儿痉挛症最为常见,部分患者还可表现为晚发型LGS,患者早期多无婴儿痉挛症的表现,并常表现为反射性癫痫[29]。其他情况如染色体结构异常,影响了重要基因的表达,也可导致LGS。
LGS缺乏明确的基因型与临床表型的对应关系,与分子遗传学相关的LGS,癫痫起病年龄较早,多在2岁以内,最初发作形式以癫痫性痉挛最常见,随后发展为多种类型发作。患者常具有较为严重的发育迟滞,表现为重度以上的智力障碍、运动系统异常如肌张力障碍、共济失调等,合并生长发育相关基因突变的患者多具有小头畸形、面部畸形等特点。患儿多社会功能低下,行为障碍和言语障碍常见。头颅核磁多正常或轻度异常。
图1呈现出每一例具有基因突变的LGS患者的起病年龄和智力障碍情况。携带离子通道相关基因、突触传递相关基因突变的LGS患者存在更为严重的智力障碍。在合并生长发育相关基因突变的LGS患者中,具有GPR56、WWOX、ARX、EEF1A等基因突变的患者可能更容易伴有严重的智力障碍。
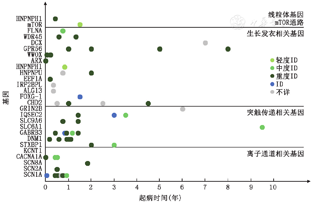
注:LGS为Lennox-Gastaut综合征;ID为智力障碍
LGS存在潜在的分子遗传学病因,可能的致病性变异多为累及单基因的新生突变,导致蛋白的功能或结构发生变化,引起离子通道的异常、突触传递的异常、神经系统发育的异常、信号传导的异常等致病。虽然越来越多的LGS相关基因被报道,但是目前针对LGS遗传学的研究缺乏对病理生理机制的深入挖掘,尚不能清楚回答众多突变基因如何导致了LGS的发生。另外,同一基因的突变可产生完全不同的表型,相似的表型可由不同的基因突变导致,因此需要进一步建立从基因型到代谢表型、蛋白质表型、临床表型甚至脑网络表型等的对应关系。最后,LGS以及EE的分子遗传学研究不仅辅助于临床诊断,更有必要应用于精准治疗,例如将离子通道阻滞剂应用于GOF突变的患者[7],再如大麻二酚通过与非大麻素类受体如GPR55、TRP通道等结合降低神经系统兴奋[30],以及应用mTOR抑制剂如依维莫司抑制mTORC1的活性,这些基于分子遗传学的精准治疗均可能有效减少癫痫发作。因此,未来应进一步在发现和总结致病基因的基础上深入机制研究、阐明多表型组学内在联系、寻找针对性治疗手段。

























