
本文报道1例新生儿Omenn综合征,表现为生后皮肤大面积蜕皮,伴有鱼鳞状脱屑,全外显子基因检测发现RAG1基因突变。Omenn综合征是一种特殊的原发性免疫缺陷病,主要表现为红皮病或鱼鳞病等皮肤改变,预后差,应早筛查、早诊断,改善预后。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿女,因“生后反应差30 min”入院。患儿系第1胎第1产,胎龄37+6周顺产娩出,出生体重3 050 g,生后反应差,呻吟吐沫,Apgar评分1 min 7分、5 min 9分,羊水Ⅲ度粪染,脐带绕颈1周。父母非近亲结婚,否认遗传及代谢病史。入院查体:双下肢皮肤大面积蜕皮,三凹征阳性,双肺呼吸音粗。胸部X线片:两肺野透过度减低。白细胞波动于(16.5~27.6)×109/L,CRP正常。入院后予呼吸支持、阿莫西林钠克拉维酸钾抗感染、丁酸氢化可的松软膏及复方多粘菌素软膏涂抹全身,皮肤仍较干燥。住院11 d白细胞计数仍增高,家属要求出院,院外未规律治疗及复查。生后22 d患儿因“发现脐部分泌物6 d”再次入院。入院查体:体重2 109 g(<P3),脐部黄色分泌物,全身蜕皮及白色脱屑,鳞屑脱落后皮肤呈红色。白细胞计数波动于(32.4~57.4)×109/L。血、尿培养阴性;脑脊液培养屎肠球菌阳性。胸部X线片:双肺野散在斑片影,右上肺条索影;右肋膈角变钝。腹部超声:肝大,脾周少量积液。免疫球蛋白:IgG 5.74 g/L,IgA <0.07 g/L,IgM 0.31 g/L。流式细胞术检测:CD4+/CD8+ 0.86,CD19+细胞百分比0.04%,余基本正常。入院后排黄稀便10余次/日,精神反应及吃奶好,口服蒙脱石散无缓解;查体发现脐炎及双侧中耳炎,予脐部消毒及外耳道冲洗;先后予美罗培南联合替考拉宁、头孢曲松联合盐酸万古霉素治疗,仍反复出现肺炎。结合患儿临床表现及免疫学检查,考虑特殊类型免疫缺陷病,行全外显子测序家系突变检查发现,患儿RAG1基因有2个杂合变异:c.2867T>C(p.I956T)变异来自母亲,该变异为已报道致病性变异;c.2686T>G(p.W896G)变异来自父亲,该变异为未见文献报道的新发变异(图1)。诊断Omenn综合征。生后第50天,家属因患儿预后差自动出院,出院后15 d患儿因严重感染死亡。
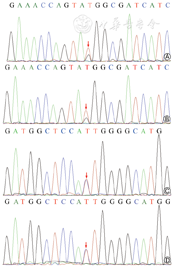
Omenn综合征是一种特殊的原发性免疫缺陷病,为常染色体隐性遗传,约占严重联合免疫缺陷病患儿的3%[1],最常见病因为RAG1或RAG2基因突变后T细胞和B细胞受体基因的基因组重排缺陷。Omenn综合征的皮肤表现可能为首发临床表现[2]。本例患儿出生时即存在皮肤大面积蜕皮,第2次入院时全身蜕皮伴有鱼鳞状脱屑,与既往报道相符。除有严重联合免疫缺陷病相似临床表现外,Omenn综合征还可表现为慢性腹泻、反复感染和生长发育迟缓,伴肝脾肿大、特殊皮肤损害(如剥脱的红皮病)、嗜酸性粒细胞增多等特殊表现,免疫学指标表现为外周T细胞数量高于正常[3, 4]。本例患儿存在反复感染、腹泻、肝大、生长发育落后,流式细胞术检测结果示T系细胞、嗜酸性粒细胞比例升高,全外显子测序家系突变筛选发现患儿RAG1基因复合杂合突变,患儿父母为携带者,但表型正常,符合常染色体隐性遗传。Omenn综合征的有效治疗方法为造血干细胞移植,但移植后病死率仍在40%左右;移植前可通过应用免疫抑制剂,如环孢素A等改善皮肤表现[5]。
所有作者均声明不存在利益冲突

























