研究组蛋白去乙酰化酶沉默信息调节因子1 (silent information regulator 1,SIRT)对心肌缺血再灌注介导的心肌细胞焦亡的影响及相关机制。
选择95只SD雄性大鼠,体重250-300 g,其中80只采用随机数字表法分为5组:假手术组、模型组、SIRT1高表达组、SIRT1低表达组和单磷酸腺苷激活的蛋白激酶(adenosine monophosphate-activated protein kinase,AMPK)信号通路抑制组,每组16只;另15只大鼠采用随机数字表法分为3组:对照组,SIRT1高表达组和SIRT1低表达组,每组5只。
80只大鼠每组处理方法如下:(1)假手术组:仅开胸暴露心脏,不做心肌缺血再灌注处理;(2)模型组:心肌内注射空白载体慢病毒7 d后,构建心肌缺血再灌注模型;(3)SIRT1高表达组:心肌内注射SIRT1高表达慢病毒7 d后,构建心肌缺血再灌注模型;(4)SIRT1低表达组:心肌内注射SIRT1小干扰RNA (small interfering RNA,siRNA)表达慢病毒7 d后,构建心肌缺血再灌注模型;(5)AMPK信号通路抑制组:心肌内注射SIRT1高表达慢病毒7 d后,给予AMPK信号通路抑制剂compound C,构建心肌缺血再灌注模型。SD大鼠麻醉后,通过慢病毒注射技术,按每只老鼠1×107 TU/ 30 μl的量,将空白载体慢病毒、过表达SIRT1慢病毒或SIRT1 siRNA慢病毒,在左心室前壁分别取5个部位进行心肌注射。在注射慢病毒后第7天,阻断左前降支冠状动脉40 min再松开120 min,构建心肌缺血再灌注模型。另外15只大鼠用于检测SIRT1慢病毒感染效率,分为:(1)对照组:心肌注射空白载体慢病毒7 d;(2)SIRT1高表达组:心肌内注射SIRT1高表达慢病毒7 d;(3)SIRT1低表达组:心肌内注射SIRT1 siRNA表达慢病毒7 d。
待构建模型后对大鼠行安乐死,取缺血区心肌组织,采用生化检测以评价血清心肌酶活性;采用Westernblot法检测焦亡相关蛋白核苷酸结合寡聚化结构域样受体蛋白3 (nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)、凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing a caspase activation and recruitment domain,ASC)、炎性半胱天蛋白酶(caspase)-1 P10、白细胞介素-1β (interleukin-1β,IL-1β)的表达,以及AMPK信号通路活性;另取缺血区组织做病理切片,采用TUNEL法检测心肌细胞凋亡率。采用SPSS 22. 0统计软件进行统计学分析,表达量的数值统计用均数±标准差( ±s)表示,组间对比采用独立样本t检验,P<0. 05为差异有统计学意义。柱状图使用Graph Pad Prism 7制作。
±s)表示,组间对比采用独立样本t检验,P<0. 05为差异有统计学意义。柱状图使用Graph Pad Prism 7制作。
与模型组相比,SIRT1高表达组CK-MB [(961. 64± 11.62)vs.(1 400. 32± 16.16),P<0. 05]、乳酸脱氢酶(lactate dehydrogenase, LDH)[(974. 46± 69.58)vs.(1 752. 97± 99.62),P<0. 05]水平明显降低;心肌细胞凋亡率显著减少[(30. 06± 6.10)vs.(69.36± 3.66),P<0.05];焦亡相关蛋白NLRP3 [(4. 60±0. 83)vs.(7. 58±0.77),P<0. 05]、ASC [(2. 90± 0.19)vs.(5. 28± 0. 59),P <0. 05]、caspase-1 P10 [(3.19±0. 20) vs.(5. 06± 1.01),P<0. 05]及IL-1β [(5. 05± 0.19)vs.(8. 32± 0.85),P<0.05]蛋白表达大量下降;AMPK通路活性增加[(3. 49± 1.05)vs.(0. 76± 0.02),P<0. 05];与SIRT1组比较,AM PK信号通路抑制组CK-MB [(1 568. 28±121.34)vs.(932. 79± 11.27),P <0.0 5]及LDH [(1 930. 75± 95.79) vs.(956. 41± 102. 83),P<0. 05]水平明显升高;心肌细胞凋亡率显著增加[(69.70± 2.52)vs.(29.28± 7. 03),P<0. 05]。
组蛋白去乙酰化酶SIRT1通过调控AMPK信号通路活性,降低焦亡相关因子的蛋白表达,缓解心肌缺血再灌注损伤。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
心肌因缺血发生组织坏死而导致的心肌梗死是引起患者猝死的严重心血管疾病,其发病率和死亡率逐年上升[1]。目前临床上治疗心肌梗死的方法包括药物治疗、冠状动脉介入治疗及冠状动脉旁路移植术等,这些对血流闭塞血管的开通方法是心肌梗死后心室重构及其功能衰竭的"治本"和基础治疗,但也是心肌缺血再灌注损伤(myocardial ischemia reperfusion injury,MIRI)的罪魁祸首[2]。
MIRI的过程涉及一系列复杂的病理过程,包括炎症反应、钙超载、细胞自噬和焦亡(pyroptosis)等,可导致广泛的心肌细胞死亡[3]。Nazir等[4]研究显示,在小鼠心肌缺血再灌注(ischemia reperfusion,I/ R)模型中心肌细胞及外周血中核苷酸结合寡聚化结构域样受体蛋白3 (nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)、炎性半胱天蛋白酶(caspase)-1、白细胞介素(interleukin, IL)-1β的表达升高。研究显示,NLRP3所诱导的细胞焦亡会加重糖尿病大鼠的MIRI,通过抑制NLRP3炎症小体,可降低在高糖和I/ R条件下培养的H9C2细胞的损伤和由细胞焦亡所引起的细胞死亡[5]。陈玉磊等[6]研究结果表明,I/ R心肌细胞中NLRP3、凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing a caspase activation and recruitment domain, ASC)、caspase-1蛋白表达升高,证明细胞焦亡参与了MIRI。因此,抗心肌细胞焦亡治疗已成为MIRI治疗的重要组成部分。
沉默信息调节因子1 (silent information regulator 1,SIRT1)是一种高度保守且依赖烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide, NAD+)、核心区域高度保守的Ⅲ类组蛋白去乙酰化酶和酰苷二磷酸(adenosine diphosphate,ADP)核糖基转移酶,可通过对组蛋白以及多种非组蛋白底物的去乙酰化作用,参与调节细胞存活、凋亡、DNA修复、细胞代谢、脂质代谢、脂肪酸氧化、氧化应激反应、胰岛素分泌、衰老和炎症反应等众多生理过程[7]。已有研究发现,在MIRI期间,SIRT1转基因小鼠体内由SIRT1介导叉头框蛋白O (forkhead box protein O,FOXO)转录因子的脱乙酰化和核易位,上调B细胞淋巴瘤2 (B cell lymphoma 2,Bcl-2)家族蛋白的Bcl-2和Bcl-xl蛋白表达,并下调促凋亡成员Bax表达,从而减轻MIRI和细胞凋亡比[8]。随后研究证实,SIRT1通过调控FOXO3α抑制MIRI介导的细胞凋亡发挥心肌保护作用[9]。NAD+可以恢复SIRT1活性、降低抑癌基因p53的乙酰化水平、减弱MIRI诱导的H9C2细胞凋亡[10]。Cattelan等[11]研究表明,SIRT1激活剂白藜芦醇通过单磷酸腺苷激活的蛋白激酶(adenosine monophosphate-activated protein kinase,AMPK)依赖性机制恢复SIRTl活性和NAD+水平,激活热休克因子-1脱乙酰化,并上调热休克蛋白表达,减少心肌细胞凋亡。这些研究表明,SIRT1在心肌梗死发生发展中发挥了非常重要的作用,其可能是防治MIRI的一个关键靶点。
综上所述,为进一步确定SIRT1抑制心肌梗死损伤的具体分子机制,本研究将在大鼠心肌I/ R动物模型上,观察SIRT1对心肌I/ R介导的焦亡相关基因表达和AMPK信号通路活性的影响,旨在揭示SIRT1降低因MIRI导致的心肌细胞焦亡的作用机制,从而为SIRT1用于治疗MIRI进一步提供科学依据。
健康成年SD雄性大鼠95只,购自中国科学院上海实验动物中心(动物合格证编号:2007000564607),清洁级,体重250-300 g。80只大鼠采用随机数字表法分为:假手术组及模型组、SIRT1高表达组、SIRT1低表达组、AMPK信号通路抑制组,每组16只。(1)假手术组:仅暴露心脏,不做I/ R模型处理。(2)模型组:心肌内注射空白载体慢病毒7 d后,构建心肌I/R模型。(3)SIRT1高表达组:心肌内注射SIRT1高表达慢病毒7 d后,构建I/ R模型。(4)SIRT1低表达组:心肌内注射SIRT1 siRNA表达慢病毒7 d后,构建I/ R模型。(5)AMPK信号通路抑制组:心肌内注射SIRT1高表达慢病毒7 d后,给予AMPK信号通路抑制剂compound C,并构建I/ R模型。另15只大鼠采用随机数字表法分为:对照组、SIRT1高表达组和SIRT1低表达组,每组5只。对照组心肌注射空白载体慢病毒7 d,SIRT1高表达组心肌内注射SIRT1高表达慢病毒7 d,SIRT1低表达组心肌内注射SIRT1 siRNA表达慢病毒7 d。
步骤一:慢病毒感染
取模型组、SIRT1高表达组、SIRT1低表达组和AMPK信号通路抑制组动物,腹腔注射水合氯醛(0.3 ml/ 100 g)麻醉动物,固定于手术台上。大鼠气管插管,利用小动物呼吸机以60次/min呼吸,吸气/呼气比为2∶1,潮气量18 ml/kg进行辅助通气。在左胸第4和第5肋骨之间暴露心脏之后,按每只老鼠1×107 TU/30 μl的量,使用30 G (gauge)针通过50 μl显微注射在左心室前壁,分别取5个部位进行心肌注射。其中,模型组心肌内注射空白载体慢病毒,SIRT1高表达组和AMPK信号通路抑制组心肌内注射SIRT1高表达慢病毒,SIRT1低表达组心肌内注射SIRT1小干扰RNA (small interfering RNA,siRNA)表达慢病毒。注射后迅速缝合胸腔。在心肌内注射慢病毒7 d后,构建I/R模型。
步骤二:心肌缺血再灌注模型建立
将注射了慢病毒的模型组、SIRT1高表达组、SIRT1低表达组和AMPK信号通路抑制组动物养殖7 d后,腹腔注射水合氯醛(0.3 ml/100 g)麻醉动物,固定于手术台上。大鼠气管插管,利用小动物呼吸机以60次/min呼吸,吸气/呼气比为2:1,潮气量18 ml/kg进行辅助通气。在左胸第4和第5肋骨之间暴露心脏后,将冠状动脉左前降支(left anterior descending,LAD)采用丝缝线结扎,可逆转阻断LAD,待心肌缺血40 min后,松开丝线再灌注120 min。假手术组:仅开胸暴露心脏,不做心肌I/R处理。
步骤三:样本取材
假手术组和构建模型后的大鼠行安乐死,收集心肌缺血区组织,一部分组织进行研磨提取蛋白进行血清心肌酶活性、焦亡相关蛋白表达和AMPK通路活性检测;一部分组织制作病理切片,进行TUNEL检测心肌细胞凋亡率分析。
SIRT1过表达慢病毒和SIRT1 siRNA慢病毒由上海吉凯基因化学技术有限公司构建。抗体NLRP3 (ab214185)、ASC (ab155970)、caspase-1 P10 (ab179515)、白细胞介素-1β(interleukin-1β,IL-1β)(ab2105)等由美国Abcam公司提供。TUNEL凋亡检测试剂盒购自瑞士罗氏公司。HX-300小动物呼吸机购自成都泰盟科技有限公司。激光共聚焦扫描显微镜是由日本Olympus株式会社生产。
血清乳酸脱氢酶(lactate dehydrogenase, LDH)和肌酸激酶同工酶(creatine kinase isoenzyme-MB,CK-MB)采用市售分析试剂盒(北京科美东雅生物技术有限公司)测定。
取大鼠心脏缺血梗死部位的组织,依次进行10%中性缓冲甲醛溶液固定、石蜡包埋及切片。将组织石蜡切片经脱蜡水化后,用0.1% Triton X-100和0. 1%柠檬酸钠溶液通透,将通透好的切片用磷酸缓冲盐溶液(phosphate buffer saline, PBS)漂洗沥干,用血清封闭30 min, PBS漂洗3次,按照比例加入末端标记酶和标记液配制成的TUNEL-FITC反应工作液,放入湿盒避光30 ℃,孵育1 h,加入DAPI染色剂结合心肌细胞核,将处理好的组织用甘油封片。细胞核染色呈绿色荧光定义为细胞染色阳性,在激光共聚焦扫描显微镜下计算10个高倍镜视野,阳性细胞除以总细胞数计算心肌细胞凋亡率(%)。
取大鼠心脏梗死部位的组织加入蛋白提取裂解液提取组织总蛋白检测NLRP3、ASC、caspase-1 P10、IL-1β、AMPK、p-AMPK蛋白表达。将加入提取液的组织进行匀浆,12 000 r/ min离心5 min,取上清液测蛋白浓度。取50 μg蛋白于4 ℃凝胶电泳,电转移1 h至硝酸纤维膜,用5%脱脂奶粉溶液4 ℃封闭过夜,加入1抗体室温孵育2 h, TTBS缓冲液洗膜3×10 min,加入辣根过氧化物酶(horseradish peroxidase, HRP)标记的2抗体室温孵育膜2 h, TFBS缓冲液洗膜4× 15 min。曝光底片,扫描保存为电脑文件,用Quantity One 4.62软件将条带的灰度值数字化。以β-肌动蛋白(β-actin)作内参照,目的条带与其相比得到相对量。
采用SPSS 22.0统计软件进行统计学分析,每只大鼠每个数据重复测量3次后取平均值。LDH、CK-MB、细胞凋亡率、NLRP3、ASC、caspase-1 P10、IL-1β蛋白及p-AMPK/ AMPK表达量的数值统计用均数±标准差( ±s)表示,假手术组和模型组、模型组和SIRT1高表达组、模型组和SIRT1低表达组、SIRT1高表达组和AMPK信号通路抑制组的组间对比采用独立样本t检验,P<0. 05为差异有统计学意义。柱状图使用Graph Pad Prism 7制作。
±s)表示,假手术组和模型组、模型组和SIRT1高表达组、模型组和SIRT1低表达组、SIRT1高表达组和AMPK信号通路抑制组的组间对比采用独立样本t检验,P<0. 05为差异有统计学意义。柱状图使用Graph Pad Prism 7制作。
慢病毒感染效率评价结果显示:用Western blot法检测质粒转染效率:对照组、SIRT1高表达组和SIRT1低表达组的慢病毒转染分别为(1. 37±0. 18)、(3. 77±0.30)和(0. 76± 0.10)(图1)。SIRT1高表达组的慢病毒转染较对照组明显升高(P<0. 05);SIRT1低表达组的慢病毒转染较对照组明显降低(P<0. 05),说明慢病毒已经有效感染至心肌。
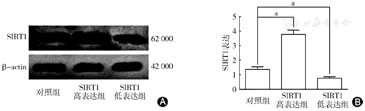
A图示Western blot检测质粒转染效率;B图示Western blot检测质粒转染结果柱状图。对照组、SIRT1高表达组、SIRT1低表达组的慢病毒转染分别为(1.37±0.18)、(3.77±0.30)和(0.76±0.10),SIRT1高表达组的慢病毒转染较做手术组明显升高;SIRT1低表达组的慢病毒转染较对照组明显降低。aP<0.05,差异有统计学意义。SIRT1:沉默信息调节因子1;β-actin:β-肌动蛋白
生化检测提示:与假手术组比较,心肌缺血40 min后再灌注120 min,模型组血清心肌酶CK-MB和LDH活性明显增加(P<0.05);SIRT1高表达组CK-MB和LDH水平较模型组明显降低(P<0.05);SIRT1低表达组CK-MB和LDH水平较模型组明显升高(P<0.05)(图2)。
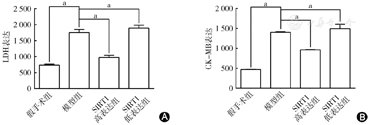
A图示LDH检测心肌损伤;B图示CK-MB检测心肌酶活性。与假手术组比较,心肌缺血40 min后再灌注120 min,模型组血清心肌酶CK-MB、LDH活性明显增加;SIRT1高表达组CK-MB、LDH水平较模型组明显降低;SIRT1低表达组CK-MB、LDH水平较模型组明显升高。aP<0.05,差异有统计学意义。SIRT1:沉默信息调节因子1;CK-MB:肌酸激酶MB;LDH:乳酸脱氢酶
心肌细胞凋亡结果显示:应用TUNEL法和激光共聚焦显微镜检测MIRI后心肌细胞的凋亡,假手术组、模型组、SIRT1高表达组、SIRT1低表达组的心肌凋亡阳性细胞分别为(2.06%±1.16%)、(69.36%±3.66%)、(30.06%±6.10%)和(69.74%±6.74%)。模型组的凋亡阳性细胞数量明显高于假手术组(P<0.05);SIRT1高表达组的凋亡阳性细胞数量明显低于模型组(P<0.05)(图3)。上述结果说明,在慢病毒有效感染至心肌,并上调SIRT1表达基础上,可有效抑制MIRI。
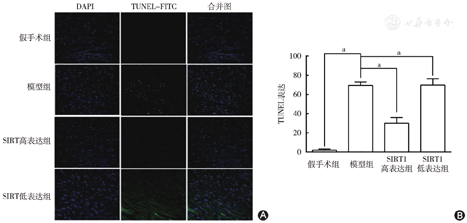
A图示TUNEL法检测MIRI后心肌凋亡阳性细胞;B图示TUNEL法检测MIRI后心肌凋亡阳性细胞结果柱状图。假手术组、模型组、SIRT1高表达组及SIRT1低表达组的心肌凋亡阳性细胞分别为(2.06%±1.16%)、(69.36%±3.66%)、(30.06%±6.10%)和(69.74%±6.74%)。模型组的凋亡阳性细胞数量明显高于假手术组;SIRT1高表达组的凋亡阳性细胞数量明显低于模型组。aP<0.05,差异有统计学意义。SIRT1:沉默信息调节因子1;MIRI:心肌缺血再灌注损伤
采用Western blot法检测焦亡相关蛋白表达,包括NLRP3、ASC、caspase-1 P10和IL-1β蛋白(图4)。结果显示,与假手术组比较,模型组NLRP3、ASC、caspase-1 P10和IL-1β蛋白表达明显升高(P<0.05);SIRT1高表达组NLRP3、ASC、caspase-1 P10和IL-1β蛋白表达较模型组明显降低(P<0.05);SIRT1低表达组NLRP3、ASC、caspase-1 P10和IL-1β蛋白表达较模型组明显升高(P<0.05)。结果表明,SIRT1可显著抑制心肌I/ R介导的焦亡相关因子表达。
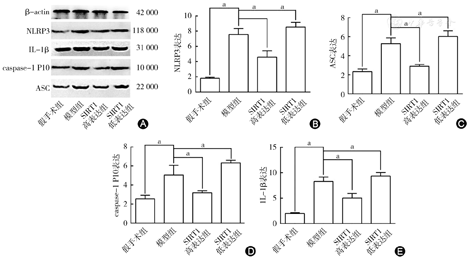
A图示Western blot法检测焦亡相关蛋白表达;B图示Western blot检测NLRP3蛋白表达结果柱状图;C图示Western blot检测ASC蛋白表达结果柱状图;D图示Western blot检测caspase-1 P10蛋白表达结果柱状图;E图示Western blot检测IL-1β蛋白表达结果柱状图。与假手术组比较,模型组NLRP3、ASC、caspase-1 P10、IL-1β蛋白表达明显升高;SIRT1高表达组NLRP3、ASC、caspase-1 P10、IL-1β蛋白表达较模型组明显降低;SIRT1低表达组NLRP3、ASC、caspase-1 P10、IL-1β蛋白表达较模型组明显升高。aP<0.05,差异有统计学意义。SIRT1:沉默信息调节因子1;NLRP3:核苷酸结合寡聚化结构域样受体蛋白3;ASC:凋亡相关斑点样蛋白;caspase:胱天蛋白酶;IL-1β:白细胞介素-1β
Western blot法检测p-AMPK/AMPK蛋白表达情况:模型组的p-AMPK/AMPK蛋白明显低于假手术组(P<0.05);感染SIRT1至心肌后,SIRT1高表达组p-AMPK/AMPK蛋白表达较模型组和假手术组明显升高(P均<0.05);SIRT1低表达组p-AMPK/AMPK蛋白表达较模型组和假手术组明显降低(P均<0.05)(图5)。结果表明,SIRT1在抑制心肌缺血再灌注损伤时激活了AMPK信号通路。
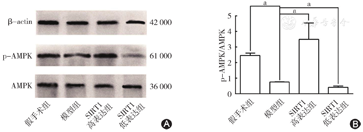
A图示Western blot法检测p-AMPK/AMPK蛋白表达;B图示Western blot法检测p-AMPK/AMPK蛋白表达结果柱状图。模型组的p-AMPK/AMPK蛋白明显低于假手术组;感染SIRT1至心肌后,SIRT1高表达组p-AMPK/AMPK蛋白表达较模型组和假手术组明显升高;SIRT1低表达组p-AMPK/AMPK蛋白表达较模型组和假手术组明显降低。aP<0.05,差异有统计学意义。SIRT1:沉默信息调节因子1;β-actin:β肌动蛋白;AMPK:单磷酸腺苷激活的蛋白激酶
生化检测提示:与假手术组比较,心肌缺血40 min后再灌注120 min,模型组血清心肌酶CK-MB、LDH活性明显增加(P<0.05);感染SIRT1至心肌后,SIRT1高表达组CK-MB、LDH水平较模型组明显降低(P<0.05);AMPK信号通路抑制组CK-MB、LDH水平较模型组明显升高(P<0.05)(图6)。
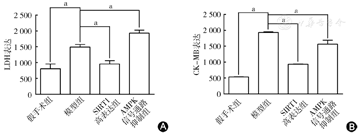
A图示LDH检测心肌损伤;B图示CK-MB检测心肌酶活性。与假手术组比较,心肌缺血40 min后再灌注120 min,模型组明显增加血清心肌酶CK-MB、LDH活性;感染SIRT1至心肌后,SIRT1高表达组CK-MB、LDH水平较模型组明显降低;AMPK信号通路抑制组CK-MB、LDH水平较模型组明显升高。aP<0.05,差异有统计学意义。SIRT1:沉默信息调节因子1;AMPK:单磷酸腺苷激活的蛋白激酶;LDH:乳酸脱氢酶;CK-MB:肌酸激酶同工酶
TUNEL法和激光共聚焦显微镜检测MIRI后心肌细胞的凋亡:假手术组、模型组、SIRT1高表达组、AMPK信号通路抑制组的心肌凋亡阳性细胞分别为2.06%±1.16%、69.36%±3.66%、30.06%±6.10%和69.74%±6.74%。模型组的凋亡阳性细胞数量明显高于假手术组(P<0.05);SIRT1高表达组的凋亡阳性细胞数量明显低于模型组(P<0.05)(图7)。结果表明,SIRT1的心肌保护作用是通过AMPK信号通路实现的。
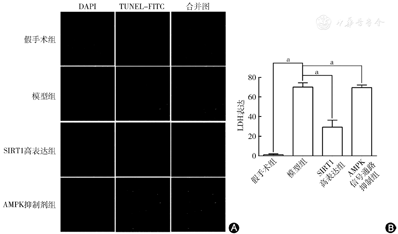
A图示TUNEL法检测MIRI后心肌凋亡阳性细胞;B图示TUNEL法检测MIRI后心肌凋亡阳性细胞结果柱状图。假手术组、模型组、SIRT1高表达组、AMPK信号通路抑制组的心肌凋亡阳性细胞分别为2.06%±1.16%、69.36%±3.66%、30.06%±6.10%和69.74%±6.74%。模型组的凋亡阳性细胞数量明显高于假手术组;SIRT1高表达组的凋亡阳性细胞数量明显低于模型组。aP<0.05,差异有统计学意义。SIRT1:沉默信息调节因子1;AMPK:单磷酸腺苷激活的蛋白激酶;MIRI:心肌缺血再灌注损伤;LDH:乳酸脱氢酶
心肌缺血是指心脏的血液灌注减少导致心肌供氧量减少,心肌能量代谢异常,不能支持心脏正常工作的一种病理状态[12]。目前的治疗方法如药物治疗、冠状动脉介入治疗、冠状动脉旁路手术等虽然可以缓解急性心肌梗死患者心肌缺血状态,但也导致了MIRI加重细胞氧化应激、炎症反应、心肌能量代谢异常等一系列病理反应,导致心肌梗死面积扩大,影响患者的预后[13]。因此,阻断MIRI后氧化应激、炎症反应进程及改善能量代谢成为改善心肌细胞焦亡的新靶点。
沉默信息调节因子(Sirtuins)家族是一类依赖于烟酰胺腺嘌呤二核氨酸(nicotinamde adenine dinucleotide,NAD+)、核心区域高度保守的组蛋白去乙酰化酶和ADP核糖基转移酶,调控细胞细胞存活、凋亡、DNA修复、细胞代谢、胰岛素分泌、衰老和炎症反应等过程[6]。哺乳动物Sirtuins家族中已发现7个同源蛋白(即组蛋白去乙酰化酶SIRT1-SIRT7),其中SIRT1与酵母Sir2的同源性最高。SIRT1基因在胎儿和成人组织中广泛表达,特别在心脏中表达相对较高[14]。以往研究发现,SIRT1通过激活转录因子FOXO1上调抗氧化基因SOD和下调促凋亡基因Bax来抑制氧化应激,从而在MIRI中起到保护心脏的作用[15]。这些研究提示,SIRT1作为有利因素,对病理性心脏起到明显保护作用。本研究发现,在构建大鼠心肌缺血40 min再灌注2 h的MIRI模型后感染SIRT1至心肌,可有效抑制大鼠血清心肌酶CK-MB、LDH水平以及心肌细胞的凋亡率。而当下调SIRT1的表达后,反而加重心肌损伤。这就表明SIRT1可减少缺血再灌注介导的心肌细胞损伤。但MIRI发生机制复杂、涉及因素较多,SIRT1对MIRI后的心肌细胞损伤的调控机制尚未完全阐明,特别是SIRT1是通过怎样的机制调控靶基因的表达,需要深入探讨。
细胞焦亡被认为是一种新的促炎程序性细胞死亡方式,由caspase-1介导,并伴有大量炎性因子及促炎因子的释放,最终诱发级联放大的炎性反应[16,17]。在细菌、病毒、细胞损伤等信号的刺激下,细胞内的NLRP3炎症小体的PRRs作为感受器,识别这些信号,通过下游的接头蛋白ASC与pro-caspase-1结合形成多蛋白质复合物,从而激活caspase-1。活化的caspase-1一方面切割gasdermin (GSDM)蛋白诱导细胞膜穿孔,释放细胞内容物,诱发炎症效应;另一方面切割IL-1β和IL-18的前体,形成具有活性的IL-1β和IL-18,募集更多的炎性因子,扩大炎症反应[18,19]。Sandanger等[20]通过结扎小鼠冠状动脉发现心肌梗死后小鼠心室肌NLRP3、IL-1β及IL-18信使RNA的表达水平明显升高。对离体鼠的心脏进行缺血再灌注处理,用caspase-1的抑制剂VX-765处理后,能降低损伤,这主要与抑制caspase-l、IL-lβ、IL-18、ACS、NLRP3、GSDM-N末端的表达有关。进一步研究发现,对已经过P2Y12受体抑制剂处理的大鼠,进行左前降支结扎再灌注前,尾静脉注射caspase-1的抑制剂VX-765后,结果不仅降低炎症与焦亡相关因子的表达,而且能减少鼠的心肌梗死面积、降低左心室重构、提高左室功能[21]。这些研究都说明,抑制焦亡反应可有效缓解心肌缺血再灌注损伤,提高心功能。
因此,SIRT1在MIRI过程中可减少心肌细胞损伤作用,这是否与调控NLRP3炎症小体活性抑制焦亡反应有关?Han等[22]在C57BL/6J小鼠体内构建心肌缺血再灌注模型后,使用SIRT1激活剂SRT1720后,通过调控PDH相关的葡萄糖氧化代谢途径抑制NLRP3炎症小体活性和焦亡反应。这一研究初步说明SIRT1可抑制NLRP3炎症小体活性缓解心肌缺血损伤。但包括Han研究在内的多篇成果显示,SRT1720主要在高血糖高血脂的相关研究具有显著疗效[23,24]。那么在单纯心肌缺血再灌注的大鼠体内,上调SIRT1能否起到同样的抑制焦亡反应效果,不得而知。为此,我们通过慢病毒感染技术,直接上调或下调大鼠心肌细胞中SIRT1的表达,发现感染SIRT1至心肌后能下调因缺血介导的NLRP3、ASC、切割后的caspase-1、IL-1β、IL-18表达。而抑制心肌细胞中的SIRT1表达后,上述NLRP3炎性小体在心肌中的表达进一步增加。这些结果都能证实,SIRT1可以很好地控制NLRP3炎症小体的活化,并阻止caspase-1的激活,抑制焦亡反应,发挥心肌保护作用。
AMPK的活性影响着机体组织的能量代谢、神经元修复、血管再生等功能。Cattelan等[25]发现白藜芦醇通过AMPK依赖性机制恢复SIRTl活性和NAD+水平,激活热休克因子-1脱乙酰化,并上调热休克蛋白表达,减少心肌细胞凋亡;而Chen等[26]的研究发现曲美他嗪可通过SIRT1- AMPK信号通路减弱败血症引起的心肌巨噬细胞促炎反应,抑制巨噬细胞促炎性反应介导的心肌细胞凋亡,提示SIRT1可通过AMPK信号通路起到心肌保护作用。本研究在确定SIRT1可抑制心肌焦亡的基础上,进一步验证SIRT1-AMPK信号通路与SIRT1抗心肌缺血再灌注介导心肌细胞损伤的相关性。结果发现:SIRT1对NLRP3炎症小体相关因子的抑制作用与其对AMPK信号通路活性的激活作用同步发生,而抑制SIRT1表达后,这一激活作用消失。这就说明SIRT1抑制焦亡反应的心肌保护作用与AMPK信号通路相关。
虽然上述结果证实了AMPK信号通路参与了SIRT1的心肌保护作用,但是AMPK是否在这一过程中发挥了主要作用,还未见报道。为确定这一疑问,我们使用特异性抑制剂Compound C抑制AMPK信号通路活性后,发现SIRT1的心肌保护作用被抑制,导致血清心肌酶活性增加、心肌细胞损伤加重、存活率降低。基于此,我们认为AMPK作为关键信号通路在MIRI过程中发挥重要作用。
综上所述,SIRT1作为有利因素,可有效减少心肌缺血再灌注介导的大鼠心肌细胞焦亡,对病理性心脏起到明显保护作用,而这一作用可能是通过激活AMPK信号通路并抑制NLRP3炎性小体活性实现的。这一发现不仅揭示了心肌梗死的病理机制,同时也为临床治疗提供了相关的理论依据。


























