
普拉德-威利综合征(PWS)是一种因为缺乏父源染色体15q11.2-q13区域相关基因的表达而引起的多系统受累的复杂遗传性疾病。其主要遗传机制有3种类型,即父源缺失型、母源单亲二倍体型和印记缺陷型。基于PWS的不同遗传机制可进行遗传咨询,对已生育该病患者的夫妇进行再次生育评估及产前诊断。PWS的致病原因及机制较为复杂,分子遗传学的快速发展及相关研究使得对该病得到了更深入的认识。现就PWS的遗传学研究进展作一综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
普拉德-威利综合征(Prader-Willi syndrome,PWS)是一种罕见的遗传性疾病,该病最早由Prader等[1]于1956年报道而得名。PWS发病原因为父源染色体上15q11.2-q13区域基因表达缺失,其致病原因及遗传机制非常特殊且复杂,绝大多数病例为新发突变,在许多人群中,PWS的发病率为1/30 000~1/10 000[2],大部分病例呈散发状态。PWS患者有着典型的面部特征,包括窄前额、杏仁眼、窄鼻梁、薄上唇、嘴角朝下等,60%~70%的患者伴有斜视;这些特征在出生时不明显,但随着时间的推移而逐渐突出。其临床表现随年龄而变化,肌张力低和吮吸无力可导致婴儿期喂养困难;儿童期开始的贪食及过量进食导致肥胖;发育迟缓、身材矮小、认知障碍、性腺发育不良和行为问题等会随年龄增长而显现出来。肥胖及其并发症是PWS患者死亡的主要原因。故PWS又称"肌张力减退-智力低下-性腺功能减退-肥胖综合征",俗称"小胖威利综合征"。PWS作为一种遗传综合征,表型随年龄不同而变化较大。充分认识PWS的遗传机制有助于该病的早期诊断和遗传咨询。
PWS为父源染色体15q11.2-q13区域印记基因的功能缺陷所致,与PWS相关的区域长约6 Mb[3]。从染色体长臂端粒至着丝粒方向可依次分为远端非印记区域、Angelman综合征(AS)印记区、PWS印记区及非印记区域(近着丝粒处断裂点BPl和BP2间)4个亚区(图1):(1)远端非印记区域含有3个GABA受体基因簇、OCA2基因、HERC2基因和常见远端断点BP3;(2)AS印记区包含优先母系表达的UBE3A和ATP10A基因;(3)PWS印记区包含仅父系表达的5个多肽编码基因(MKRN3、MAGEL2、NECDIN、双顺反子SNURF-SNRPN、C15orf2),一组核仁小RNA(snoRNAs)以及一些反义转录本(包括UBE3A的反义转录本);(4)2个常见的近端断点(BP1和BP2)之间的近端非印迹区域包含4个双表达基因,即NIPA1、NIPA2、CYF1P1和TUBGCP5[4]。
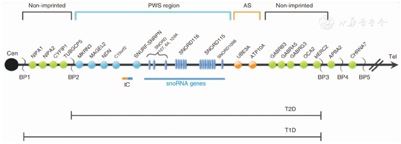
注:PWS:普拉德-威利综合征;AS:Angelman综合征;T1D:1型缺失;T2D:2型缺失 PWS:Prader-Willi syndrome;AS:Angelman syndrome;T1D:type 1 deletion;T2D:type 2 deletion
其中,位于PWS印记区的MAGEL2基因是黑色素瘤抗原基因家族(melanoma antigen gene family,MAGEs)的一员,编码1 249个氨基酸,介导蛋白间相互作用。MAGE蛋白作为泛素连接酶调节因子,可识别并结合具有RING结构域的E3(E3 RING)泛素连接酶,形成MAGE-RING连接酶(MRL)稳定复合物(图2)[5],MAGE蛋白可增强RING E3泛素连接酶的活性并改变其定位,使其识别其他底物,从而改变细胞信号通路[6]。MAGEL2是一种母系印记、富含GC的单个外显子基因,仅由未甲基化的父本等位基因表达[6]。
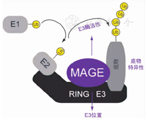
注:MAGE:黑色素瘤抗原基因 MAGE:melanoma antigen gene
已有研究表明,MAGEL2在下丘脑中高表达[7],其在介导膜蛋白从核内体回收过程中发挥重要作用[5]。已有报道膜蛋白通过逆转运复合体(retromer)途径回收,包括管家蛋白(如阳离子非依赖性甘露糖-6-磷酸受体,CI-M6PR),信号蛋白[如Wntless和G蛋白偶联受体(GPCRs)]和细菌毒素[8]。MAGEL2、TRIM27 E3泛素连接酶和USP7去泛素化酶共同组成一个多亚基蛋白复合物(MAGEL2-USP7-TRIM27,MUST),MUST通过泛素化激活WASH肌动蛋白成核促进因子,促进retromer回收膜蛋白[4]。
目前已在2种Magel2基因敲除小鼠模型的研究中,初步阐明了MAGEL2的生理功能。小鼠Magel2基因定位于小鼠7号染色体上的人PWS簇的同源区域,也是母系印记[9]。第一种小鼠模型是用LacZ报告基因替换Magel2编码序列,而Magel2启动子不变[10]。这种模型的胎鼠病死率略有增加,新生小鼠喂养困难,早期表现出生长迟缓,断奶后体质量增加,伴随着代谢和内分泌功能障碍(包括下丘脑功能障碍)而出现肥胖[10]。此外,各种神经递质水平也有所下降,包括5-羟色胺、多巴胺等,并表现出一些异常行为,如新环境中出现焦虑等[11,12]。第二种小鼠模型是敲除Magel2启动子和大部分编码区(CDS区),仅保留最后的1 165 bp[13]。这些Magel2敲除小鼠表现出较高的新生鼠病死率,最突出的神经内分泌功能障碍是催产素产生障碍。新生小鼠的高病死率和行为缺陷可以通过催产素注射来治疗[13,14]。以上研究表明MAGEL2有着非常重要的细胞生物学功能。
印记中心(imprinting center,IC)位于PWS印记区内SNURF-SNRPN基因启动子区域,掌控印记区内父源印记与母源印记之间的转换。正常人的母源染色体15q11.2-q13区域中SNRPN基因的CpG岛高度甲基化,而父源染色体SNRPN的CpG岛未甲基化,因此母源基因失活,而父源SNRPN基因表达(遗传印记)。当父源15q11.2-q13区域中的SNRPN缺失(或功能缺陷)时,患者即表现出PWS的表型。
已有研究表明,在PWS患者和PWS父本缺失的小鼠模型中,常染色质组蛋白赖氨酸N-甲基转移酶-2(EHMT2,也称为G9a)的2种选择性抑制剂UNC0638和UNC0642,可以激活PWS区SnoRNA簇中的SNORD116候选基因的母本拷贝[15]。
通过重亚硫酸盐基因组测序进一步分析发现,UNC0642和UNC0638降低了PWS-IC区组蛋白H3赖氨酸9的二甲基化(H3K9me2),而未改变PWS-IC中的DNA甲基化。选择具有优良药代动力学特性的UNC0642对父本缺失的小鼠模型进行治疗,发现UNC0642可以改善新生小鼠的存活和生长情况,提供了一个基于表观遗传学的PWS治疗的证据[15]。
PWS发病的遗传学机制主要有3个:父源缺失、母源单亲二倍体(UPD)和印记中心缺陷(图3)。
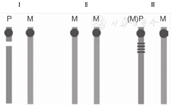
注:Ⅰ :父源缺失;Ⅱ:母源单亲二倍体;Ⅲ:印记缺陷 Ⅰ:deletion of the paternal critical region;Ⅱ:maternal uniparental disomy;Ⅲ:imprinting center defect
大多数PWS患者是由于父源15q11.2-q13区域的微缺失所致。在PWS患者中,父系缺失占65%~75%[16]。大多数父系缺失的患者都有2个常见的近端断点之一(BP1或BP2)和1个常见的远端断点(BP3)。染色体15q11.2-q13区域容易缺失是因为在这些断裂点(BP1、BP2和BP3)附近有重复序列的多重拷贝,会在减数分裂的时候造成非同源配对和异常重组,导致缺失(引起PWS或AS取决于来自父源还是母源)、重复(母源或父源)、三倍体及15号染色体倒位[17]。
UPD是指2条15号染色体都来源于母亲,没有一条来自父亲。在PWS患者中,UPD占20%~30%。UPD已被证实与高龄产妇有关[16]。大多数UPD的产生大致可概括为减数分裂过程中发生突变的二体配子与正常配子受精结合形成三体合子。此后,卵裂过程中发生染色体后期迟滞将其中1条丢失,三体变成二体。如果丢失的是来自正常配子的那条染色体,而保留在合子里的2条染色体就是同二体,三体合子变成单亲二体,这个过程被称为"三体营救"。基于多例在胎盘细胞中发现三体而在胚胎细胞中发现UPD报道,可知"三体营救"是形成UPD型PWS的主要原因[18,19]。此外,配体互补及有丝分裂复制都可导致单亲二倍体的产生,但发生的概率很低。
印记中心缺陷是影响15号染色体的印记过程。在PWS患者中,印记缺陷占1%~3%。大多数印记缺陷是由表观遗传因素引起的。尽管存在双亲等位基因,但只有母体的DNA是甲基化模式。在这些基因中DNA序列没有发生改变,它们被认为是精子形成过程中,或是在罕见的体细胞嵌合体的早期胚胎中发生随机错误而没能清除母源印记[20]。此外,大约在15%的有印记缺陷的患者中发现位于印记中心区域的SNRPN基因启动子的5′末端存在缺失(7.5~>100 kb)[21]。其中,大约有一半的遗传是来自未受影响的父亲且他的母源遗传15号染色体上印记中心发生缺失;另一半是新生的在精子形成或受精后发生的15号染色体父系遗传缺失[22,23]。
基于PWS的不同分子遗传机制(表1)进行遗传咨询,可对已生育PWS患者的夫妻进行再次生育评估[4]。几乎所有15q11.2-q13的缺失都是新发5~6 Mb缺失(Ⅰa),再发风险非常低(<1%)。在父源缺失的PWS患者中可以用荧光原位杂交(FISH)进行核型分析,在极少数的情况下,缺失是由于染色体重排(Ⅰb)导致的。UPD15是典型的新发突变(Ⅱa),再发率小于1%,但若在父母双方中存在罗伯逊易位则概率上升,因此在先证者中进行核型分析是必要的。印记缺陷的PWS患者应该由有检测经验的实验室对他们进行印记中心缺失检测。大约85%的印记缺陷是新发表观遗传突变(Ⅲb)且再发风险<1%,其余15%是基于印记中心的微缺失(Ⅲa)。
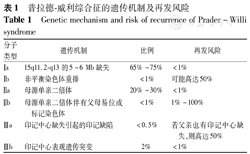
普拉德-威利综合征的遗传机制及再发风险
Genetic mechanism and risk of recurrence of Prader-Willi syndrome
普拉德-威利综合征的遗传机制及再发风险
Genetic mechanism and risk of recurrence of Prader-Willi syndrome
| 分子类型 | 遗传机制 | 比例 | 再发风险 |
|---|---|---|---|
| Ⅰa | 15q11.2-q13的5~6 Mb缺失 | 65%~75% | <1% |
| Ⅰb | 非平衡染色体重排 | <1% | 可能高达50% |
| Ⅱa | 母源单亲二倍体 | 20%~30% | <1% |
| Ⅱb | 母源单亲二倍体伴有父母易位或标记染色体 | <1% | 1%~100% |
| Ⅲa | 印记中心缺失引起的印记缺陷 | <0.5% | 若父亲也有印记中心缺失,则高达50% |
| Ⅲb | 印记中心表观遗传突变 | 2% | <1% |
由表1可知,PWS的再发风险与其印记基因缺陷的类型有关。大多数家庭再发风险低于1%,而某些病因再发风险会高达50%,甚至达到100%,如母亲15号染色体罗伯逊易位等。进行再发风险评估有助于确定PWS患儿是否发生了新发突变,或是否有如嵌合体等其他诱发因素。当父母进行检测时,亲代荧光原位杂交术(FISH)或微阵列比较基因组杂交(aCGH)研究都能够为父母双方提供准确的遗传咨询、预后指导及再发风险评估,如果有必要的话,建议父母双方进行生殖选择。
PWS相关的染色体区域涉及基因印记、可变剪接变异、基因拷贝数变异等遗传机制,其复杂性导致一系列涉及多器官多系统的复杂表型。为提高PWS患者的生活质量与寿命,需对其进行遗传检测以明确其致病机制,从而方便医师评估疾病进展。对生育过PWS患儿的夫妇也应进行相应的遗传咨询与产前诊断。此外,还应根据患者的具体情况制定个性化的管理方案。对于PWS患者进行基因检测及临床信息收集非常重要,这有助于对PWS及疑似患者进行进一步的诊断、遗传检测和疾病进展评估。目前已知PWS的遗传机制是来自染色体15q11-q13的父本表达基因的缺失,同时母本染色体该区域中的相同基因在结构上完整但被甲基化抑制了其表达,然而PWS的确切分子机制尚未完全阐明,有待进一步揭示。由于基因组印记缺陷疾病具有独特的分子和表观遗传特征,对其进行深入研究将为探索人类表观遗传疾病的诊断和治疗带来希望。
所有作者均声明不存在利益冲突

























