
分析2个齿状核红核苍白球路易体萎缩症(DRPLA)家系的临床和遗传学特点,总结基因型与表型的相关性。
收集2018年7月至2019年3月就诊于北京大学第一医院2个以遗传性癫痫和共济失调为主要表现的家系中先证者和相关家系成员的外周血、临床资料和辅助检查结果,通过采用全外显子组测序及毛细管电泳和片段分析检测三核苷酸(CAG)重复次数,对先证者及其家系成员进行基因检测,并对2个家系中所有受累家系成员进行临床和遗传特点分析。
2个家系符合DRPLA诊断,共11例受累者,均表现为智力运动倒退,其中7例有癫痫发作(包括肌阵挛发作、局灶性发作和全面强直阵挛发作等)。在同一家系中不同受累者间临床表现差异较大,子代较亲代发病年龄更早,病情更重。起病年龄最小者2岁,最大者45岁,其中儿童期起病者5例。11例受累者中,已有5例死亡,死亡原因包括癫痫发作、可疑癫痫性猝死(SUDEP)和病情进展等。6例存活的受累者发现ATN1基因第5外显子CAG重复次数异常。家系2中先证者的爷爷临床表现正常,但也存在ATN1基因第5外显子CAG异常重复,可能为中间等位基因。
DRPLA的主要临床特点为癫痫发作、共济失调和智力倒退,存在遗传早现现象。儿童期发病者少见,常以癫痫发作为首发症状就诊。该病预后不良,早期明确诊断有助于指导遗传咨询。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
齿状核红核苍白球路易体萎缩症(dentatorubral-pallidoluysian atrophy,DRPLA)是一种少见的神经遗传病,最早由Naito和Oyanagi[1]报道,临床表现存在异质性,即使同一个家庭的受累者临床表型轻重不同。20岁之前起病的患者通常表现为进行性肌阵挛癫痫(progressive myoclonic epilepsy,PME),临床特征包括癫痫发作、共济失调、肌阵挛和进行性认知倒退、痴呆等[2,3]。DRPLA为常染色显性遗传,由编码多聚谷氨酰胺片段的ATN1基因第5外显子三核苷酸(CAG)重复序列不稳定扩增导致[4,5]。需通过毛细管电泳分离实验和片段分析进行检测并计算CAG重复次数。该病诊断需结合家系临床特点和基因检测结果。目前国内仅有少数关于DRPLA家系的报道,且儿童期发病的仅有7例患者[5,6,7,8]。本研究分析2个DRPLA家系的临床和遗传特点,并总结基因型与表型的相关性。本研究2个家系中共11例受累者,其中5例为儿童期起病。现报告如下。
2018年7月至2019年3月就诊于北京大学第一医院儿科,基因诊断明确的2个DRPLA家系。参与本研究的患儿监护人均知情同意,并签署知情同意书。本研究通过医院医学伦理委员会批准[批准文号:2012(453)]。
收集先证者和相关家系成员的外周血各4 mL、临床资料和辅助检查结果。对2个以遗传性癫痫和共济失调为主要表现的家系,采用全外显子组测序(WES)未发现致病基因者,进一步通过毛细管电泳和片段分析法进行CAG重复次数检测,以明确致病基因,该部分实验由赛福解码基因科技有限公司协助完成。根据DRPLA基因第5外显子CAG重复及其前后的序列进行引物设计,采用荧光标记M13末端加尾法进行PCR,PCR产物进行琼脂糖凝胶电泳,并通过毛细管电泳分离试验和CEQ 8000核酸分析仪进行片段分析。DRPLA基因荧光标记M13末端加尾法PCR、毛细管电泳分离实验及片段分析见参考文献 [9]。
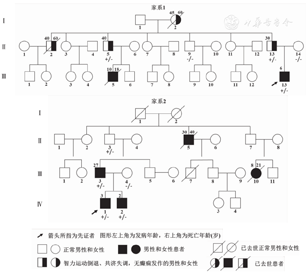
本研究中2个DRPLA的家系图见图1。临床特点见表1,共11例受累者。家系1先证者(Ⅲ-13)为6岁7个月出现癫痫发作,癫痫发作类型包括肌阵挛发作、局灶性发作和全面强直阵挛发作(GTCS)。患儿发病前运动发育正常,语言表达较同龄儿稍差,计算能力差,3个月竖头稳,6个月独坐,1岁独走,1岁2个月会叫爸爸妈妈。发病后智力运动倒退,7岁出现走路不稳,进行性加重,13岁时仅能卧床,末次随访15岁,卧床不能行走,仅会说单字。查体患儿头围52 cm,四肢肌张力低下,膝腱反射活跃,双眼上视困难,水平运动无受限。其堂哥10岁(Ⅲ-5)起病,有癫痫发作,具体表现不详,18岁睡眠中去世,可疑癫痫性猝死(sudden unexpected death in epilepsy,SUDEP)。先证者的奶奶(Ⅰ-2)、大伯(Ⅱ-2)、二伯(Ⅱ-5)、父亲(Ⅱ-13)均有走路不稳,伴认知倒退,起病年龄30~45岁,且均无癫痫发作。先证者的奶奶和大伯分别于60岁去世,具体原因不详。本家系在本课题组进行性肌阵挛癫痫(PME)相关研究中已有报道[6]。
2个DRPLA家系11例患者临床表型和基因检测结果
The clinical features and genetic testing results of 11 patients in two DRPLA families
2个DRPLA家系11例患者临床表型和基因检测结果
The clinical features and genetic testing results of 11 patients in two DRPLA families
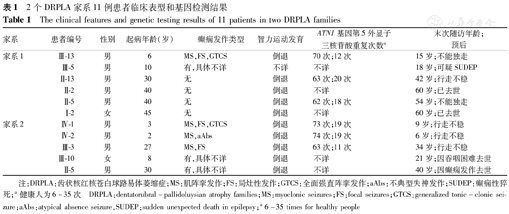
| 家系 | 患者编号 | 性别 | 起病年龄(岁) | 癫痫发作类型 | 智力运动发育 | ATN1基因第5外显子三核苷酸重复次数a | 末次随访年龄;预后 |
|---|---|---|---|---|---|---|---|
| 家系1 | Ⅲ-13 | 男 | 6 | MS,FS,GTCS | 倒退 | 70次;12次 | 15岁;不能独走 |
| Ⅲ-5 | 男 | 10 | 有,具体不详 | 不详 | 不详 | 18岁;可疑SUDEP | |
| Ⅱ-13 | 男 | 30 | 无 | 倒退 | 63次;20次 | 42岁;行走不稳 | |
| Ⅱ-2 | 男 | 40 | 无 | 倒退 | 不详 | 60岁;已去世 | |
| Ⅱ-5 | 男 | 40 | 无 | 倒退 | 62次;18次 | 54岁;不能独走 | |
| Ⅰ-2 | 女 | 45 | 无 | 倒退 | 不详 | 60岁;已去世 | |
| 家系2 | Ⅳ-1 | 男 | 3 | MS,FS,GTCS | 倒退 | 73次;19次 | 9岁;行走不稳 |
| Ⅳ-2 | 男 | 2 | MS,aAbs | 倒退 | 74次;19次 | 6岁;行走不稳 | |
| Ⅲ-3 | 男 | 27 | MS,FS | 倒退 | 63次;11次 | 34岁;行走不稳 | |
| Ⅲ-10 | 女 | 8 | 有,具体不详 | 倒退 | 不详 | 21岁;因吞咽困难去世 | |
| Ⅱ-5 | 男 | 30 | 有,具体不详 | 倒退 | 不详 | 40岁;因癫痫发作去世 |
注:DRPLA:齿状核红核苍白球路易体萎缩症;MS:肌阵挛发作;FS:局灶性发作;GTCS:全面强直阵挛发作;aAbs:不典型失神发作;SUDEP:癫痫性猝死;a健康人为6~35次 DRPLA:dentatorubral-pallidoluysian atrophy families;MS:myoclonic seizures;FS:focal seizures;GTCS:generalized tonic-clonic seizure;aAbs:atypical absence seizure,SUDEP:sudden unexpected death in epilepsy;a 6-35 times for healthy people
家系2先证者(Ⅳ-1)为3岁开始出现癫痫发作,癫痫发作类型包括肌阵挛发作、局灶性发作和全面强直阵挛发作(GTCS)。患儿起病前运动发育正常,语言发育迟缓,1岁2个月独走,8岁开始出现走路不稳,易跌倒,伴智力倒退,会说5~6字短句。查体患儿头围52 cm,四肢肌张力低下,膝腱反射活跃,无明显眼球运动障碍。先证者的弟弟(Ⅳ-2)2岁出现发育落后,6岁时出现癫痫发作,癫痫发作类型包括肌阵挛发作和不典型失神发作。先证者的父亲(Ⅲ-3)为27岁出现癫痫发作,癫痫发作类型包括肌阵挛发作和局灶性发作,随后出现行走不稳。先证者爷爷的弟弟(Ⅱ-5)为30岁出现癫痫发作,具体表现不详,30岁后出现智力倒退和行走不稳,40岁因癫痫发作去世。患儿的堂姑(Ⅲ-10)8岁出现癫痫发作,具体表现不详,14~15岁出现运动倒退,21岁因病情进展去世。
家系1的先证者和父亲均进行4 h的脑电图监测和头颅影像学检查。先证者的脑电图显示广泛性棘波、棘慢波发放及局灶性放电,其父亲显示为偶见额极、额、中央区不典型尖波散发。先证者及其父亲均有脑沟加深和小脑萎缩(图2)。
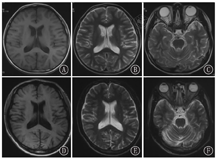
家系2的先证者、弟弟及父亲进行4 h的脑电图监测,均显示广泛性棘波、棘慢波发放及局灶性放电,先证者的脑电图见图3。先证者进行头颅影像学检查,提示大脑皮质和小脑萎缩。
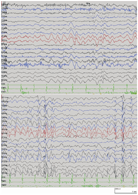
注:背景显示双侧枕区少量、低-中波幅4~5 Hz θ活动;发作间期显示双侧半球较多广泛性中至高波幅棘波、棘慢波放电,以及后头部局灶性放电 The background shows a few low-to-medium amplitude 4-5 Hz θ activities in the bilateral occipital region;the interical period shows mid-high amplitude spikes,spike and waves spikes discharge in bilateral hemispheres,and focal discharges in the occipital area
2个家系的先证者及父母WES检测 均未发现明确致病性基因突变,进一步分析2个家系的临床特点,受累者临床表现包括癫痫发作、智力运动倒退和小脑共济失调,且子代起病年龄均早于亲代,临床症状也更重,存在遗传早现的现象。提示2个家系为遗传性脊髓小脑共济失调(spinocerebellar ataxia,SCA)并癫痫发作,SCA中较常见的突变形式是CAG/多核苷酸的重复扩增,通过基于荧光标记的毛细管电泳和片段分析法对先证者和父母进行SCA常见基因的动态突变检测,随后再对其他受累者进行验证。
基因检测结果显示,2个家系中所有存活的受累者均存在ATN1基因第5外显子CAG重复次数异常,具体见表1。家系1的先证者(12次和70次)、父亲(20次和63次)、二伯(18次和62次);家系2的先证者(19次和73次)、弟弟(19次和74次)和父亲(11次和63次)。对2个家系中表型正常者进行验证,家系1先证者母亲(13次和14次)、三伯(11次和19次);家系2先证者母亲(18次和19次)CAG重复次数正常,而家系2先证者爷爷CAG重复次数异常,为8次和56次。
DRPLA首次于1982年在一个日本家庭中报道[1]。该病由ATN1基因第5外显子CAG重复序列的不稳定扩增导致,健康人中该片段的重复数为7~23,DRPLA患者中可扩增至49~75,且重复片段的长度与起病年龄呈负相关,与疾病严重程度呈正相关[10]。起病年龄越早的患者更倾向于表现为PME表型[3]。文献报道,根据起病年龄,本病可分为少年型和成年型[8]。其中起病年龄<20岁为少年型,通常有进行性肌阵挛、癫痫发作、进展性共济失调、智力倒退。成年型又分为早发型和晚发型,40岁以前起病的早发型以共济失调为主,可伴有痴呆和癫痫发作;40岁以后起病的晚发型表现为共济失调、记忆力下降等。本研究中2个家系中20岁以前起病的患者5例,均有癫痫发作及智力运动倒退、共济失调表现,其中3例进行基因检测,CAG重复序列的重复次数均≥70次;40岁及以后起病的患者3例,均表现为运动倒退伴认知障碍,而无癫痫发作,其中1例患者进行基因检测,CAG重复次数为62次;20~40岁起病的患者3例,均有运动倒退伴认知障碍,其中2例合并癫痫发作,其中2例进行基因检测,CAG重复次数均为63次。基因检测结果与其对应的起病年龄、临床表现均与Shahwan等[3],朱蔚文等[8],Kälviäinen[10]报道一致。
国内有关DRPLA家系的报道尚少,张爱娟等[11]报道的最大1个家系为5代共85人中有14例受累者,其中11例接受基因检测,均存在ATN1基因第5外显子CAG异常重复。DRPLA属于SCA中的一种疾病,有研究对827例共济失调患者进行临床特点和致病基因进行研究,仅发现3例DRPLA患者,均存在ATN1基因第5外显子CAG异常重复[9]。国内报道的DRPLA患者中,儿童期发病的仅有7例[5,6,7,8]。该病临床少见,在儿童中更为罕见。
本研究的2个家系中均有多位受累者,家系WES检测均未发现明确致病性基因,进一步分析家系临床特点,先证者均为10岁前出现癫痫发作和智力运动发育倒退,且先证者的起病年龄较父辈更早、临床表现较父辈更重,即2个家系均存在遗传早现的现象,提示2个家系临床特点符合SCA合并癫痫发作。通过基于荧光标记的毛细管电泳和片段分析法对先证者进行SCA常见基因的动态突变检测,发现2个家系中受累者均存在ATN1基因第5外显子CAG重复次数异常,表型正常者为正常。结合临床症状和基因检测结果,诊断该2个家系均为ATN1基因突变导致的DRPLA。本病中20岁以前起病的患者需与其他常见的可导致PME的疾病进行鉴别,如神经元蜡样质脂褐质沉积症、Unverricht-Lundborg病、Lafora病、戈谢病、尼曼匹克病、肌阵挛癫痫伴破碎样红纤维等[12]。20岁之后起病的患者需与亨廷顿病及其他SCA相鉴别[8]。
曾有文献报道1个DRPLA家系中,先证者哥哥20岁,ATN1基因CAG重复次数为53次,无临床症状;另1个家系先证者的父亲64岁,ATN1基因CAG重复次数为52次,无临床症状[13]。分析该现象,可能由于ATN1基因CAG重复次数尚处于过渡区,即中间等位基因,这类成员可能出现临床症状,也可能终生表型正常,但中间等位基因在传代过程中容易发生进一步的扩增,子代倾向于发病[13,14,15]。本研究中家系2先证者的爷爷(Ⅱ-3)ATN1基因CAG重复次数为56次,但临床表型正常,属于中间等位基因;但先证者父亲(Ⅲ-3)ATN1基因CAG重复次数为63次,27岁发病,表现为癫痫发作和智力运动倒退;先证者及其弟弟ATN1基因CAG重复次数分别为73次和72次,分别于3岁和2岁发病,表现为癫痫发作和智力运动发育倒退。
本研究中2个DRPLA家系的临床和遗传特点研究提示,当一个家系中存在多个受累者,临床表现包括癫痫发作、小脑共济失调和智力倒退且存在遗传早现时,如果家系全外显子组测序未能发现致病基因时,应进一步采用毛细管电泳和片段分析法进行基因检测以明确诊断。
所有作者均声明不存在利益冲突

























