
1岁11月龄患儿以腹胀、呕吐起病,临床表现为腹腔高压综合征、重度营养不良,腹部影像学检查提示肠梗阻,剖腹探查见所有肠道严重扩张,未见机械性梗阻,回肠造瘘术后仍有腹胀,后全外显子组基因检测提示为ACTG2基因c.769C>T; p.Arg257Cys突变,诊断为原发性内脏肌病致假性肠梗阻。该病临床罕见,预后差,目前以对症支持治疗为主。当患儿出现顽固性难治性腹胀且排除机械性肠梗阻时,需考虑此病,早期基因检测可明确诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
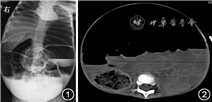
患儿 女,1岁11月龄,因“腹胀、呕吐20 d,加重2 d”于2020年7月收入山东第一医科大学附属省立医院小儿重症医学科。入院前20 d患儿出现腹胀,伴呕吐,2~3次/d,呕吐物为少量胃内容物,非喷射性,未予特殊处理;入院前15 d呕吐好转,仍有腹胀,后于外院治疗,腹胀无缓解;入院前2 d腹胀较前加重,遂至山东第一医科大学附属省立医院门诊,立位腹X线片示肠管明显扩张并积气,可见多个阶梯状气液平面(图1),查血常规及肝功生化示血小板降低(84×109/L)、钾降低(1.84 mmol/L),拟诊“肠梗阻、低血钾、血小板减少症”收住院。患儿发病以来,精神尚可,进食量较前减少,尿量可,排便1~2次/d,为少量黄色稀便。患儿系其母第2胎第2产,足月经剖宫产出生,出生体重4.2 kg,母亲孕期体健,否认生后窒息缺氧史,生后体重增长缓慢。父、母及兄均体健。家族史无特殊。
入院体格检查:体温37.0 ℃,脉搏133次/min,呼吸31次/min,血压 80/50 mmHg(1 mmHg=0.133 kPa),身长82 cm,头围47.5 cm,体重9.0 kg。神志清,精神欠佳,体形消瘦,全身皮肤黏膜干燥,弹性差,双侧眼窝凹陷,口唇干燥,呼吸急促,三凹征阳性,腹部明显膨隆,腹围69 cm,腹壁静脉显露,未见肠型及蠕动波,无压痛反跳痛及肌紧张,全腹叩诊呈鼓音,移动性浊音阴性,肠鸣音消失,四肢肌张力略低,四肢末梢凉,毛细血管再充盈时间>5 s,余未见异常。
实验室检查(括号内为参考值范围):动脉血气分析示pH 7.53(7.35~7.45),氧分压52 mmHg(95~100 mmHg),二氧化碳分压58 mmHg(35~45 mmHg);血常规示白细胞计数7.7×109/L(4.0×109~10.0×109/L)、淋巴细胞0.321(0.200~0.400)、中性粒细胞0.583(0.500~0.700)、血红蛋白112 g/L(120~160 g/L)、血小板计数206×109/L(150×109~350×109/L)、C反应蛋白4.2 mg/L(0~8.0 mg/L)、降钙素原0.21 μg/L(0~0.05 μg/L);血乳酸1.40 mmol/L(0.44~1.78 mmol/L);肝功能示天冬氨酸转氨酶 59 U/L(8~40 U/L)、丙氨酸转氨酶38 U/L(5~40 U/L)、白蛋白32 g/L(40~55 g/L)、钙 1.84 mmol/L (2.25~2.58 mmol/L)、磷 0.44 mmol/L(1.29~1.94 mmol/L)、镁 0.85 mmol/L(0.56~0.76 mmol/L)、二氧化碳结合力 23.7 mmol/L(22.0~31.0 mmol/L)、钾2.3 mmol/L(3.5~5.5 mmol/L)、钠125.6 mmol/L(135.0~145.0 mmol/L)、氯85.1 mmol/L(95.0~105.0 mmol/L)、乳酸脱氢酶 394 U/L(120~250 U/L);尿蛋白+,尿潜血弱阳性;粪潜血弱阳性;甲状腺功能无异常;结缔组织病相关检查无异常。
影像学检查:腹部CT示腹盆部明显膨隆,胃腔及部分肠管明显扩张并积气,可见多个阶梯状气液平面,腹腔肠管拥挤,结构显示不清。肝脏大小、形态可,胆囊、胰腺显示不清。脾脏及双肾平扫未见明显异常密度灶。膀胱空虚,内见导尿管影。子宫显示不清。腹盆腔未见明显增大淋巴结及明显积液影(图2)。
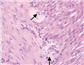
诊疗经过:入院后初步诊断为腹胀原因待查(肠道发育畸形?低钾性肠麻痹?),休克,呼吸衰竭观察(Ⅱ型),重度脱水,电解质紊乱,血小板减少症,重度营养不良。患儿存在休克表现,给予积极扩容、抗休克、抗感染、禁食、胃肠减压、静脉营养等对症支持治疗。患儿呼吸急促无缓解,给予呼吸机辅助呼吸,多次复查立位腹X线片均可见肠管扩张并积气,存在多个气液平面。经治疗后,患儿腹胀仍进行性加重,呼吸困难无缓解,血乳酸进行性增高,提示出现腹腔高压综合征。入院后第5天急症行剖腹探查术及肠道减压术,并送检外周血进行全外显子组基因检测。外科术中见所有小肠、结肠广泛严重扩张,以横结肠扩张最为显著,肠壁增厚质韧,呈皮革状,未见肠蠕动,探查肠道未见机械性梗阻部位。给予回肠袢式造瘘,术中分别取乙状结肠、回肠、横结肠及阑尾送病理活检,术后返回重症监护室。肠道活检病理结果示乙状结肠、回肠肌层神经丛查见神经节细胞,数量不一,部分伴发育不良;横结肠肌层神经丛内查见少量神经节细胞,部分伴发育不良;阑尾纵行肌层外缘近浆膜处可见神经丛,查见神经节细胞,部分伴发育不良(图3)。根据病理结果,考虑暂不能排除先天性巨结肠同源病,该病目前无特殊治疗,以对症支持为主。患儿术后给予禁食、胃肠减压等处理,但腹胀缓解不明显,胃肠减压引流量始终较多,回肠造瘘口处引流量少,多次复查腹立位X线片仍可见肠管明显扩张并积气。入院后第20天全外显子组基因结果示ACTG2基因(OMIM: 102545)c.769C>T; p.Arg257Cys杂合突变,导致第257位的精氨酸被半胱氨酸所取代,美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics, ACMG)指南表明该突变具有致病性[1],可导致常染色体显性遗传疾病——内脏肌病(OMIM:155310),患儿父母ACTG2基因均未发现突变(图4),其兄体健,家长拒绝为其行基因检测。至此该患儿诊断明确,为ACTG2基因突变引起的内脏肌病,该病可引起慢性假性肠梗阻(chronic intestinal pseudo obstruction,CIPO)。家长放弃治疗,出院2个月后因病情加重死亡。
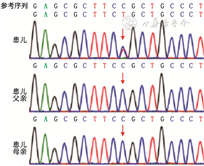
原发性内脏肌病是引起CIPO的原因之一,包括家族型内脏肌病和散发型内脏肌病,前者表现为常染色体显性或隐性遗传,后者病因不明或与某些病毒感染有关。病变多位于小肠,也可累及结肠或食管[2, 3, 4]。本例患儿小肠及结肠均受累,为ACTG2基因c.769C>T; p.Arg257Cys的杂合突变,为常染色体显性遗传。
ACTG2基因编码平滑肌的γ2肌动蛋白,突变后阻止肌动蛋白聚合,降低了平滑肌细胞的收缩力,可引起常染色体显性家族型内脏肌病,可累及肠道、膀胱和(或)子宫,临床上可表现为CIPO、巨膀胱-细小结肠-肠蠕动不良综合征、先天性腹肌缺如综合征、肠旋转不良、喂养不耐受、复发性尿路感染、膀胱功能障碍以及胆囊炎或胆石症等[5, 6]。研究显示超过40%的CIPO患者是因为ACTG2基因突变所致,且均为错义突变,但尚未有研究表明基因型和表型具有相关性[5, 7]。突变形式包括新发型及家族遗传型,前者自婴儿期即开始出现喂养不耐受,肠旋转不良及肠梗阻的表现,预后差;而后者则于青春期或成年期出现CIPO的表现,常需进行手术切除扩张的肠段,最终需要依赖胃肠外营养,可存活至高龄[5]。本例患儿家族中未发现其他突变病例,考虑为ACTG2基因的新发突变。
ACTG2基因突变所致内脏肌病的病理改变差别迥异,据报道异常的病理表现包括全肠道固有肌层重度萎缩,平滑肌细胞排列紊乱且肿胀、γ2肌动蛋白阳性的细胞内包涵体减少,平滑肌肌动蛋表达减少,结缔组织减少并且存在大神经节,S100免疫染色显示肌间神经丛紊乱;也有研究报道远端回肠和盲肠全层活检正常[6]。本例患儿仅见乙状结肠、回肠及阑尾肌层神经丛的神经节细胞数量不一,部分伴发育不良,符合内脏肌病的病理改变,但该病理表现也可见于先天性巨结肠同源病,基因诊断可进行鉴别[8]。
ACTG2基因突变引起的内脏肌病所致CIPO临床罕见,诊断困难,疑诊时应先排除机械性肠梗阻,并排除引起假性肠梗阻的其他原因,如结缔组织病(如系统性红斑狼疮、进行性系统性硬化)、肠道神经丛病变以及电解质紊乱等,明确诊断需依靠基因检测。本例患儿入院后积极对症支持治疗,腹胀仍进行性加重,剖腹探查排除机械肠梗阻,并排除了其他原因,最终基因检测明确诊断。但本病尚无有效的治疗方法,急性期应禁食、胃肠减压、纠正电解质紊乱、促进胃肠蠕动。通常不建议手术治疗,因ACTG2基因编码的肌动蛋白功能障碍,若行外科手术,可能导致吻合失败;仅对于病变局限、保守治疗无效、出现肠穿孔等并发症且有条件行胃肠外营养者可考虑外科手术,极重症患者可考虑肠移植。该病预后因突变类型不同,新发型突变者预后差,而家族遗传型突变者依靠胃肠外营养可存活至高龄[5, 9]。
所有作者均声明不存在利益冲突

























