
本文报道1例TEMPI综合征的诊治过程。患者47岁男性,出现继发性红细胞增多伴有促红细胞生成素升高,血IgG κ单克隆丙种球蛋白31.75 g/L,骨髓见16%的异常浆细胞,查体颜面、胸及背部可见毛细血管扩张,腹部CT可见左肾小低密度影,考虑为肾周积液。除不存在肺内分流,患者的临床症状及实验室检查符合TEMPI综合征的诊断标准。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者男,47岁,因“发现红细胞升高30余月”于2017年2月13日入院。患者既往高血压10余年;吸烟20年,20支/d;间断饮酒20年,250 g/d。患者2014年8月因阑尾炎急性发作前往当地医院。血常规示:白细胞(WBC)4.9×109/L,红细胞(RBC)6.87×1012/L,血红蛋白(Hb)217 g/L,血小板(PLT)136×109/L。促红细胞生成素(EPO)279.76 mU/ml(2.59~18.50 mU/ml)。血红蛋白电泳、PNH克隆检测、甲状腺功能、抗核抗体、ENA抗体谱及肿瘤标志物均正常。骨髓病理:造血组织粒红系增生,红系增生明显。融合基因P190、P210、JAK2 EXON12、JAK2 V617F均阴性。当地诊断为“真性红细胞增多症”。予以“干扰素”注射治疗1个月后,略有缓解,后患者间断行放血治疗(2014年1次、2015年2次、2016年2次),放血治疗后1个月红细胞继续上升,血红蛋白达180 g/L左右。患者间断感头晕、头胀,查体:颜面潮红,颜面、胸及背部可见毛细血管扩张(图1),余均阴性。为求进一步诊治来本院。
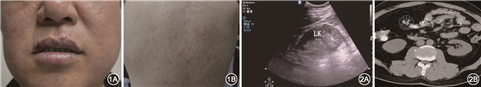
入院后检验结果,血常规:WBC:5.52×109/L,RBC:7.53×1012/L,Hb:146 g/L,PLT:297×109/L, 平均红细胞血红蛋白量(MCH)19.4 pg,平均红细胞体积(MCV)70.3 fL,红细胞平均血红蛋白浓度(MCHC) 276 g/L。EPO 253.38 mU/ml。血气分析正常。血管内皮生长因子(VEGF)357.7 μg/L(0~142 μg/L)。血β2微球蛋白:3.11 mg/L↑。免疫球蛋白G (IgG)49.3 g/L↑,轻链κ定量4 790 mg/dl↑,轻链λ定量178 mg/dl。血免疫固定电泳:在γ区可见一条单克隆IgGκ成分,单克隆免疫球蛋白(M蛋白)计算为31.75 g/L。尿免疫固定电泳、蛋白电泳阴性。骨髓细胞学检查:幼稚浆细胞4%,成熟浆细胞12%。骨髓病理:骨髓增生极度活跃,浆细胞比例升高(10%~20%),限制性表达kappa。骨髓流式:异常细胞群占有核细胞的2.97%,表达CD138、CD38、CD56、cKappa,不表达CD117、CD19、cLambda,为异常浆细胞表型。染色体核型正常。荧光原位杂交技术(FISH):RB-1基因缺失阳性,IgH基因重排阳性,CCND1/IGH阳性,TP53、1q21、1p32未见异常。二代测序:NOTCH2 Exon23 49.42%;NOTCH1 Exon21 48.29%。泌尿系B超:左肾体积增大,右肾体积正常,包膜规整,肾实质回声增强;皮质与髓质界限欠清。腹部CT:双肾形态、大小正常,边缘毛糙,左肾门上方水平紧邻肾皮质可见小低密度影,部分突出肾轮廓外(图2)。
患者红细胞增多伴有EPO升高、存在单克隆丙种球蛋白及异常浆细胞,伴有毛细血管扩张及肾周积液,笔者认为这是与最近提出的TEMPI综合征相符的罕见病例之一。参考既往文献,建议其接受硼替佐米为基础的诱导治疗,必要时行自体干细胞移植,患者表示暂不同意,后回当地未行特殊治疗。2019年12月电话随访,患者述一般状况良好,面部及胸背部毛细血管扩张症仍可见,头晕乏力等不适症状有所缓解。
TEMPI综合征于2011年被首次提出[1],其特征表现为五联征:毛细血管扩张、EPO升高和红细胞增多症、单克隆丙种球蛋白病、肾周积液和肺内分流。考虑到异常浆细胞在该病中可能的作用机制,2016年WHO修订的造血和淋巴组织肿瘤分类将该病和POEMS综合征一并纳入到浆细胞疾病伴副肿瘤综合征的名下[2]。迄今为止,全世界仅有十几例病案报道[1, 3, 4, 5, 6, 7, 8, 9],发病中位年龄48岁(35~65岁),男女占比相当。
多数患者以红细胞增多症起病,易被误诊为真性红细胞增多症(PV),可通过以下三点加以鉴别:(1)几乎所有的PV患者都有获得性JAK2 V617突变,但TEMPI综合征患者一般无;(2)PV患者EPO水平一般正常,而TEMPI综合征患者的一般很高;(3)TEMPI综合征的骨髓活检通常无特殊之处,而PV等慢性骨髓增殖性肿瘤通常有特殊形态如特征性巨核细胞增生、网状蛋白纤维化或窦内造血等[10]。本例患者虽有红细胞增多,但JAK2 V617F、JAK2 EXON12突变均阴性、血清EPO水平显著增高、骨髓无典型慢性增殖性肿瘤的特征,因而考虑为伴EPO升高的继发性红细胞增多症。
本患者另一大特征为骨髓中存在异常浆细胞(16%),血清存在M蛋白(IgGκ 31.75 g/L),无CRAB和SLiM症状,诊断为冒烟型骨髓瘤(SMM)。TEMPI综合征通常以IgGκ型M蛋白为主[1, 3, 7, 9],肿瘤负荷低,血清M蛋白少(中位数11.45 g/L,5.06~36.00 g/L),骨髓中异常浆细胞通常<10%,符合意义未明单克隆免疫球蛋白血症诊断标准。也有报道和本例患者一样,浆细胞比例>10%,达到SMM诊断标准[4]。TEMPI综合征还可出现呼吸困难、肾周积液、腹痛腹胀、肾功能不全等表现,但一般晚于红细胞增多症(7~10年后)[1],本例患者虽未能证实存在肺内分流,可能是因为患者处于疾病早期所致,且既往也有报道氧分压正常的患者[5, 6]。
既往文献报道的TEMPI综合征患者对浆细胞病样治疗均有应答[1, 3, 4, 5, 6, 7, 8, 9],完全缓解率约70%。目前推荐硼替佐米为基础的治疗方案作为首选诱导治疗方案,疗程数一般6~8个周期,若评价疗效为部分缓解,则建议行自体造血干细胞移植,对于复发/难治的患者,推荐免疫调节剂或CD38单抗等其他抗浆细胞肿瘤药物。
所有作者均声明不存在利益冲突

























