
探讨1例涉及FOXG1基因的14q12q13.1缺失患儿的临床及分子生物学特征。
分析患儿的临床表现,对其进行外周血染色体核型分析和单核苷酸多态性微阵列(single nucleotide polymorphism array,SNP-array)检测。分析FOXG1相关疾病基因型与表型的相关性。
患儿为男性,出生后第8天出现喂养困难,表现为吸吮无力,同时出现下肢抖动、额部青紫,磁共振成像显示双侧侧脑室明显增宽、胼胝体发育不良。染色体核型为46,XY,del(14)(q12q13.1),SNP-array检测显示患儿存在14q11.2q13.1区9.6 Mb的缺失,涉及FOXG1等基因。
对大脑发育异常并具有运动、认知、语言障碍等的患者,应警惕FOXG1基因的拷贝数变异,及早进行SNP-array检测以明确诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
FOXG1基因位于染色体14q12区,为剂量敏感基因,参与调控神经元细胞的增殖、分化、迁移定位和凋亡等,在端脑从胚胎期至成年的发育过程中发挥重要作用。 FOXG1基因编码G1叉头蛋白,其变异可能影响G1叉头蛋白的合成及功能,影响胎儿大脑的正常发育,使患儿出现一些典型的症状,如脑结构异常、严重的躯体异常与发育障碍等。近期研究证实,FOXG1基因变异与严重智力低下、语言缺失及癫痫等多种神经发育异常相关。FOXG1基因点变异或累及FOXG1的14q12微缺失主要表现为获得性小头畸形、严重认知运动障碍、语言障碍、癫痫及刻板动作等[1,2,3,4]。
涉及FOXG1的14q12区微缺失非常罕见,全球仅报道约16例。这些缺失的临床表现与神经发育障碍以及Rett综合征(Rett syndrome,RTT)存在重叠,但其大脑影像和一些特征如运动障碍、缺乏消退性和缺乏呼吸性心律失常等又与经典的RTT有所不同[3]。除RTT外,涉及FOXG1基因的14q12微缺失还与14q12微重复存在明显的表型重叠[5],这类疾病被统称为FOXG1综合征[3]。我们报告1例涉及FOXG1基因的14q12 9.6 Mb杂合缺失,并对FOXG1基因相关疾病的临床及遗传学特征进行了分析。
患儿,男,系第4胎,足月顺产,出生时哭声洪亮,无窒息抢救史。出生第8天出现喂养困难,表现为吸吮无力,同时出现下肢抖动,额部青紫。查体:未触及前囟、颅缝重叠,眼距宽,右下肢肌张力高,足底扁平、外翻。磁共振成像提示双侧侧脑室增宽明显、胼胝体发育不良。心脏B超提示卵圆孔未闭,怀疑为21-三体综合征,随后转入我院就诊。患儿父母签署了知情同意书,本研究通过了医院伦理委员会的审查(KS-2018-KY-36)。
无菌抽取患儿外周静脉血3 mL,肝素抗凝,常规进行淋巴细胞培养、G显带。用Axio lmager Z2 Ikaros全自动扫片系统扫描,用MetaClient软件分析,按照人类细胞遗传学国际命名体制(ISCN 2009)以及卫生部发布的《胎儿染色体异常的细胞遗传学产前诊断技术标准》进行描述核型。
用试剂盒(德国Qiagen公司)提取羊水细胞基因组DNA,用美国Affymetrix公司生产的CytoScan 750k Array进行检测,用Chromosome Analysis Suite(ChAS)软件对结果进行分析。查询DGV(http://dgv.tcag.ca/dgv/app/home)、DECIPHER(http://decipher.sanger.ac.uk/)、OMIM( http://omim.org/)、UCSC(http://genome.ucsc.edu/)和PubMed( http://ncbi.nlm.nih.gov/pubmed)等数据库对结果进行判读。
2.1 G显带染色体分析显示患儿核型为46,XY,del(14)(q12q13.1),见图1。同时进行的定量荧光PCR(quantitative fluorescent-PCR,QF-PCR)检测结果亦排除了13、18、21三体。患儿父母拒绝接受染色体检查。
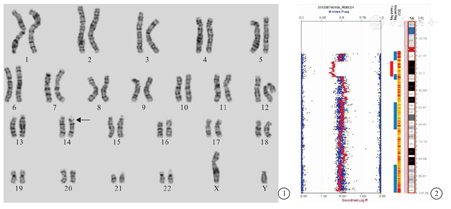
2.2 为进一步明确缺失的来源和大小,我们对患儿的外周血DNA进行了SNP-array检测,结果为arr[hg19]14q12q13.1(25 084 632-34 690 056)×1,即14q12q13.1区存在9.6 Mb的缺失(图2),涉及FOXG1、KIAAA133、PRKD1、SCFD1、STRN3 HECTD1、ARHGAP5、AKAP6等10余个功能基因。
本例患儿通过外周血染色体检查及QF-PCR检测,排除了21-三体综合征的可能性,随后SNP-array检测结果提示14q12q13.1区存在约9.6 Mb的缺失。文献报道类似片段缺失患者的主要表现包括严重发育迟缓、生长停滞、小头畸形、特殊面部(包括眼距异常、内眦赘皮、额头倾斜、低位耳、圆眉、小口及长人中等),磁共振成像可见胼胝体发育不全[6]。其缺失片段涉及FOXG1、KIAAA133、PRKD1、SCFD1、STRN3 HECTD1、ARHGAP5、AKAP6等10余个基因。其中的FOXG1为端脑转录因子,与小头畸形和胼胝体发育不全密切相关。
STRN3参与调控细胞分化和成熟。HECTD1是一种E3泛素连接酶,与脊神经缺陷有关。ARHGAP5则与前脑半球融合、心室形状、视杯形成以及大脑皮质的分层有关。
FOXG1基因的异常可导致其下游基因表达水平的改变,从而导致端脑发育缺陷[7]。 Foxg1纯合敲除鼠表现为大脑皮质减小、神经元分层异常,并于胚胎期死亡[7]。 Foxg1杂合敲除鼠具有Foxg1单倍剂量不足的缺陷,表现为脑体积变小、皮质变薄及海马、杏仁核功能损害等[7,8]。
本例患儿的症状与文献报道极为相似,其缺失片段所涉及的基因的功能也与之高度相关。14q12q13.1片段的缺失应是其患病的原因。与FOXG1基因变异或拷贝数变异相关的一组疾病统称为FOXG1相关疾病[1,2],呈常染色体显性遗传,男女均可发病,人群发生率不详。尽管文献报道的多为散发病例,但由于嵌合现象的存在,家族性病例并不罕见[9,10,11,12]。FOXG1相关疾病的共同表型主要包括早发的发育迟缓、全面性肌张力低下、智力低下、语言匮乏、癫痫、手刻板动作及孤独症谱系障碍等。不同变异类型的临床表型也有一定的差异。FOXG1基因点变异或拷贝数缺失者往往还合并获得性小头畸形、睡眠障碍及运动障碍。头颅影像学检查常有胼胝体发育不良、简单脑回及额叶白质容积减少等阳性发现,此外涉及FOXG1的14q12的大片段缺失还可有颅面部畸形[11];而FOXG1拷贝数重复者头颅影像学大多正常,多无中枢性运动障碍,少数可合并小头畸形及轻微的非特异性面容异常[13]。
目前对于FOXG1基因相关疾病的治疗仍以对症为主,强调个体化治疗,对于精神运动发育落后者应及早发现并实施康复干预,重视语言和社交能力的训练,注意癫痫、睡眠障碍等的发生,并进行适当的药物治疗。本病更重要的是预防。鉴于嵌合现象的存在,当FOXG1相关疾病的家庭再次生育时,无论亲代是否携带相应变异,均应进行产前诊断。
所有作者均声明不存在利益冲突

























